Sindrom Angelman
Lisa Vogel belajar jurnalisme jabatan dengan fokus pada perubatan dan biosains di Universiti Ansbach dan memperdalam pengetahuan kewartawanannya dalam peringkat sarjana dalam maklumat dan komunikasi multimedia. Ini diikuti oleh latihan dalam pasukan editorial Sejak September 2020 dia menulis sebagai wartawan bebas untuk
Lagi catatan oleh Lisa Vogel Semua kandungan diperiksa oleh wartawan perubatan.Sindrom Angelman (Happy Puppet Syndrome) adalah penyakit genetik yang jarang berlaku. Ia menampakkan dirinya, antara lain, melalui batasan mental dan fizikal, gangguan perkembangan (terutama bahasa) dan hiperaktif. Yang menarik ialah penampilan seperti boneka dan ekspresi wajah gembira mereka yang terkena. Ketahui lebih lanjut mengenai penyakit yang jarang berlaku!
Kod ICD untuk penyakit ini: Kod ICD adalah kod yang diiktiraf di peringkat antarabangsa untuk diagnosis perubatan. Mereka boleh didapati, misalnya, dalam surat doktor atau sijil ketidakupayaan untuk bekerja. Q93

Gambaran ringkas
- Apa itu Sindrom Angelman? Penyakit genetik yang jarang berlaku yang menampakkan dirinya melalui batasan mental dan fizikal dalam perkembangan anak
- Gejala: ciri wajah seperti anak patung, gangguan perkembangan, gangguan koordinasi, tidak ada atau hampir tidak ada perkembangan bahasa, kecerdasan berkurang, kejang, ketawa tidak berasas, ketawa, kelonggaran berlebihan, melambaikan tangan dengan gembira
- Punca: kecacatan genetik pada kromosom 15
- Diagnosis: antara lain perbualan, pemeriksaan fizikal, pemeriksaan genetik
- Terapi: Tidak ada terapi kausal; Sokongan seperti fisioterapi, terapi pertuturan, terapi pekerjaan; mungkin ubat untuk melegakan simptom (mis. jika berlaku sawan)
- Ramalan: jangka hayat normal; tidak mungkin hidup berdikari
Sindrom Angelman: Definisi
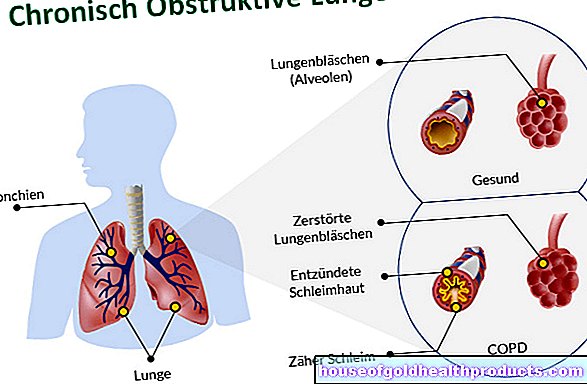
Sindrom Angelman (AS) disebabkan oleh kecacatan genetik pada kromosom 15. Kecacatan ini mengganggu perkembangan fizikal dan mental mereka yang terjejas. Gangguan perkembangan pertuturan, ketidakamanan motor, dan wajah gembira adalah gejala sindrom Angelman yang paling ketara.
Nama "Angelman Syndrome" berasal dari penemu penyakit ini, pediatrik Inggeris Harry Angelman. Pada tahun 1965, dia membandingkan gambar klinikal tiga kanak-kanak yang mempunyai ciri-ciri wajah seperti anak patung. Anak-anak banyak ketawa dan melakukan pergerakan tersentak - seperti marionettes Ini membawa nama Inggeris "Happy Puppet Syndrome" (boneka gembira).
Sindrom Angelman berlaku pada kedua-dua jantina. Risiko penyakit ini adalah sekitar 1: 20,000. Ini menjadikan sindrom penyakit yang jarang berlaku.
Sindrom Angelman: Gejala
Semasa kelahiran, kanak-kanak dengan sindrom Angelman adalah normal. Gangguan perkembangan motorik dan kognitif hanya menjadi semakin ketara pada bayi dan awal kanak-kanak. Ciri-ciri gangguan genetik adalah:
- kelewatan pengembangan motor
- terjejas koordinasi
- selalunya tidak atau hampir tidak ada perkembangan bahasa
- kecerdasan menurun
- tingkah laku hiperaktif, bersemangat tinggi
- ketawa tidak berasas
- Ketawa
- isyarat gembira (mis. melambaikan tangan)
- air liur berlebihan
- kerap melekat keluar dari lidah
Beberapa kanak-kanak dengan sindrom Angelman juga mempunyai:
- Microcephaly (kepala kecil yang tidak normal) - bukan semasa lahir, tetapi ketika ia berkembang
- Kejang
- Perubahan aktiviti otak elektrik
- kulit dan mata yang sangat ringan kerana penurunan pigmentasi (hipopigmentasi)
- Squint (strabismus)
Sindrom Angelman: punca
Penyebab sindrom Angelman adalah kecacatan genetik pada kromosom 15: fungsi gen UBE3A terganggu pada mereka yang terjejas. Gen ini biasanya menghasilkan enzim yang terlibat dalam memecah protein yang rosak atau berlebihan pada sel. Oleh itu, sel ini dapat berfungsi dengan normal.
Gen UBE3A terletak di kawasan kromosom 15q11q13. Di sana gen tertakluk pada apa yang dikenal sebagai "pencetakan genom". Ini bermaksud bahawa mereka hanya aktif pada salah satu kromosom ibu bapa (di sel badan kita terdapat dua salinan setiap kromosom - satu dari ibu dan satu dari bapa). Ini diatur oleh proses kimia - metilasi: kumpulan metil yang melekat pada titik tertentu dapat menghidupkan atau mematikan gen.
Gen ini aktif pada kedua kromosom dalam banyak sel badan, tetapi tidak pada sel saraf otak: pada banyak orang di sana, gen UBE3A pada kromosom ayah 15 dimatikan dengan mencetak. Akibatnya, UBE3A hanya aktif pada kromosom ibu 15 di otak. Ini juga bermaksud: Sekiranya salinan gen ibu menunjukkan kesalahan, ini tidak dapat dikompensasi oleh salinan gen ayah yang tidak digunakan. Dan tepatnya kombinasi ini berlaku dalam sindrom Angelman: segmen gen ayah dimatikan, segmen gen ibu rosak.
Kesalahan genetik yang mendasari boleh terdiri daripada pelbagai jenis:
- Penghapusan: Kira-kira 75 peratus daripada semua orang dengan sindrom Angelman kekurangan kawasan yang relevan 15q11q13 dengan gen UBE3A pada kromosom ibu 15. Kerana wilayah yang sesuai pada kromosom 15 bapa "dimatikan" dengan mencantumkan, tubuh dapat menggunakan enzim yang rancangan Pembinaannya disimpan dalam Gen UBE3A, jangan buat.
- Mutasi dalam gen UBE3A: Perubahan gen yang berlaku secara spontan menyebabkan maklumat yang terkandung di dalamnya hilang. Ini berlaku bagi lima hingga sepuluh peratus orang dengan sindrom Angelman. Dalam setiap kes kelima terdapat mutasi keluarga: Di sini ibu sudah melakukan perubahan genetik pada kromosom ayahnya.
- Dua kromosom ayah 15: Orang yang terjejas mewarisi kedua kromosom 15 dari bapanya, tidak ada yang berasal dari ibu (secara perubatan disebut sebagai "disomi uniparental bapa 15"). Oleh itu, tidak ada gen UBE3A yang aktif. Ini berlaku untuk sekitar satu hingga dua persen dari semua pesakit sindrom Angelman.
- Ralat pencetakan: Gen UBE3A pada kromosom ibu 15 - seperti gen kromosom ayah 15 - dimatikan dengan mencetak. Sebagai tambahan, bahagian kromosom tertentu boleh hilang (penghapusan). Kesalahan mencetak terdapat pada satu hingga empat peratus kes sindrom Angelman.
Dalam baki sepuluh hingga 15 peratus kes, penyebab sindrom Angelman tidak diketahui. By the way: jika gen ibu dimatikan dan gen ayah rosak, anak-anak menderita sindrom Prader-Willi yang disebut.
Adakah Sindrom Angelman turun temurun?
Secara amnya, risiko berulang dalam sindrom Angelman rendah. Ini bermaksud risiko bahawa ibu bapa anak yang terkena mempunyai anak lain yang juga mengalami sindrom. Walau bagaimanapun, dalam kes individu, risiko ini sangat bergantung pada kecacatan genetik yang mendasari sindrom Angelman.
Sebagai contoh, dalam sindrom Angelman disebabkan oleh dua kromosom bapa 15 (disomi uniparental bapa 15) ia kurang daripada satu peratus. Sebaliknya, sindrom Angelman kerana kesalahan mencetak dengan kehilangan segmen gen tertentu (penghapusan IC) boleh berlaku pada separuh daripada semua kes pada saudara kandung.
Risiko yang meningkat ini juga berlaku dengan mutasi UBE3A - dengan syarat ibu sudah menjadi pembawa kecacatan genetik (sekitar 20 peratus daripada semua kes mutasi). Dalam kes seperti itu, ibu mewarisi mutasi dari ayahnya. Oleh itu, UBE3A diubah dalam kromosom ibu dari ibu. Sekiranya ini dimatikan, ibu tidak akan menyedari adanya mutasi. Namun, dia dapat menyebarkan kromosom kepada anak-anaknya, di mana ia - kemudian sebagai kromosom ibu - boleh menyebabkan sindrom Angelman.
Secara teorinya, pesakit dengan sindrom Angelman dapat membiak. Bergantung pada kapan perubahan kromosom kausal terjadi (mis. Sudah pada masa perkembangan sel kuman atau segera setelah persenyawaan), risiko kadang-kadang sangat tinggi (hingga 100 persen) sehingga mereka yang terkena penyakit ini akan menularkan penyakit ini. Walau bagaimanapun, terdapat kekurangan data yang boleh dipercayai mengenai perkara ini. Terdapat kes terpencil pada bulan September 1999: Ibu, yang menghidap sindrom Angelman, menularkan penyakit ini di sini.
Sindrom Angelman: diagnosis
Sekiranya anda melihat simptom yang dijelaskan di atas pada anak anda, pakar pediatrik adalah titik kontak pertama. Dia dapat menyekat kemungkinan penyebabnya dengan lebih tepat dan merujuk anda dan anak anda kepada pakar sekiranya perlu.
orang anamnese
Langkah pertama dalam diagnosis adalah sejarah perubatan yang menyeluruh. Doktor akan menanyakan pelbagai soalan mengenai anak anda, seperti:
- Apa perubahan yang anda perhatikan pada anak anda?
- Adakah anak anda mempunyai keluhan fizikal?
- Bolehkah anak anda duduk?
- Adakah anak anda meraih objek?
- Adakah anak anda bercakap?
- Adakah anak anda sering kelihatan ceria atau teruja?
- Adakah anak anda ketawa dalam situasi yang tidak sesuai, contohnya ketika mereka kesakitan?
Pemeriksaan fizikal
Ini diikuti dengan pemeriksaan fizikal. Pakar pediatrik menguji sejauh mana kanak-kanak itu kerap mengembangkan kemahiran motor dan mental. Latihan sederhana digunakan untuk tujuan ini: Contohnya, kanak-kanak harus menumpukan perhatian pada mainan atau meraih blok bangunan secara selektif. Doktor juga memperhatikan ekspresi wajah anak. Ketawa yang kerap, ciri seperti anak patung, dan air liur adalah tanda-tanda sindrom Angelman.
Sekiranya, setelah pemeriksaan fizikal, terdapat kecurigaan penyakit yang jarang berlaku, doktor akan merujuk anda kepada pakar neurologi dan ahli genetik manusia.
Ujian genetik
Ujian genetik adalah bahagian penting dalam diagnosis sindrom Angelman. Doktor memerlukan sampel kecil sel kanak-kanak, yang dapat diperolehnya dari mukosa mulut, misalnya dengan mengambil sampel darah atau dengan menggunakan swab. Bahan genetik (DNA) sel-sel ini atau kawasan kromosom yang relevan 15q11q13 kemudian diperiksa dengan lebih terperinci di makmal.
Pada langkah pertama, para doktor memperhatikan corak metilasi segmen kromosom (analisis / ujian metilasi). Ujian lebih lanjut pada sampel yang sama (analisis penghapusan, analisis mutasi) membantu menentukan penyebab sindrom Angelman dengan lebih dekat. Untuk ini mungkin juga perlu diperiksa susunan genetik ibu bapa. Dengan cara ini, doktor dapat menentukan sama ada terdapat kecacatan genetik di sana.
Siasatan lanjut
Pemeriksaan lanjutan sering berguna. Sebagai contoh, EEG dapat digunakan untuk mengesan perubahan dalam aktiviti otak elektrik, seperti yang sering terjadi pada sindrom Angelman. Pemeriksaan oftalmologi juga boleh ditunjukkan.
Sindrom Angelman: Terapi
Sindrom Angelman tidak dapat disembuhkan - penyebab genetik penyakit ini tidak dapat diatasi. Walau bagaimanapun, campur tangan awal boleh memberi kesan positif kepada perkembangan motor dan mental mereka yang terjejas. Fisioterapi biasa, misalnya, sangat membantu. Ia dapat meningkatkan kemahiran motorik kanak-kanak, mengurangkan pergerakan terhad dan membantu mencegah penyakit sekunder seperti kecacatan tulang belakang. Kanak-kanak dengan sindrom Angelman juga mendapat manfaat daripada kaedah terapi lain seperti terapi pekerjaan dan terapi pertuturan.
Di samping itu, beberapa gejala dan keadaan yang berkaitan dengan sindrom Angelman mungkin memerlukan rawatan yang disasarkan. Sebagai contoh, ubat antikonvulsan (ubat anti-epilepsi) membantu melawan kejang, dan ubat penenang (penenang) membantu dengan gangguan tidur yang teruk.
Di laman web Verein Angelman e.V. anda akan menemui banyak maklumat mengenai sindrom Angelman, laporan pengalaman dan kejadian bagi mereka yang terjejas serta orang yang dihubungi untuk mereka yang terjejas di semua wilayah di Jerman.
Sindrom Angelman: kursus penyakit dan prognosis
Tahun pertama kehidupan
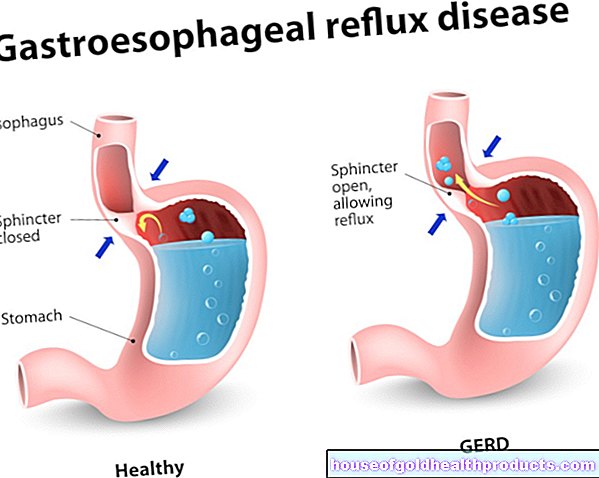
Bayi dengan sindrom Angelman cenderung menghadapi masalah penyusuan, penyusuan, dan menelan. Mereka sering menjulurkan lidah atau banyak air liur. Di samping itu, kanak-kanak dengan sindrom Angelman sering meludah (yang sering disalah anggap sebagai intoleransi makanan atau penyakit refluks). Muntah yang kerap boleh menyebabkan penurunan berat badan yang berbahaya.
Pada usia tiga hingga enam bulan, kanak-kanak dengan sindrom Angelman sering mula tersenyum. Mereka sering tertawa dan sering berdengkur dan sering menjelirkan lidah semasa kegembiraan itu.
Perkembangan motor yang tertunda biasanya dapat dilihat antara usia 6 dan 12 bulan: kanak-kanak tidak merangkak atau duduk. Pergerakan bahagian atas badan sering goyah. Ini seterusnya menjadikan duduk lebih sukar.
Sebilangan kecil mereka yang terkena sudah mengalami sawan pada usia 12 bulan.
Satu hingga tiga tahun
Dalam tiga tahun pertama kehidupan, gangguan perkembangan sindrom Angelman menjadi sangat jelas. Kanak-kanak lebih banyak mengalami sawan ringan. Sebahagian daripada mereka hiperaktif, terlalu bersemangat dan selalu bergerak. Banyak yang cenderung meletakkan tangan atau mainan di mulut mereka sepanjang masa, atau menjulurkan lidah mereka dengan kerap dan air liur. Sekiranya anak-anak sangat teruja, mereka akan sering ketawa berlebihan dan melambai dengan tangan terentang.
Perkembangan bahasa yang terganggu menjadi jelas buat pertama kalinya pada usia ini. Anak-anak "mengoceh" pada diri mereka sendiri atau menjerit dan memekik, bagaimanapun, hanya dapat mengucapkan beberapa atau tidak ada kata yang mudah dimengerti dan biasanya menggunakannya tanpa konteks.Tetapi mereka memahami bahasa dan ada juga interaksi sosial dengan orang lain.
Akil baligh dan dewasa
Pubertas sering tiga hingga lima tahun lewat pada kanak-kanak dengan sindrom Angelman. Walau bagaimanapun, kematangan seksual kemudian berkembang secara normal. Masih belum ada perkembangan bahasa jika pemahaman bahasa sering hadir. Kejang pada masa dewasa biasanya dapat diatasi dengan baik dengan ubat-ubatan.
Orang dengan sindrom Angelman mempunyai jangka hayat yang normal. Kehidupan berdikari tidak mungkin berlaku kerana keterbatasan mental.
Tags.: bayi kecil khasiat terapi




-kopfsache.jpg)