Sindrom Marfan
Mareike Müller adalah penulis bebas di jabatan perubatan dan penolong doktor untuk pembedahan saraf di Düsseldorf. Dia belajar perubatan manusia di Magdeburg dan memperoleh banyak pengalaman perubatan praktikal semasa dia tinggal di luar negara di empat benua yang berbeza.
Lebih banyak mengenai pakar Semua kandungan diperiksa oleh wartawan perubatan.Sindrom Marfan (MFS) adalah penyakit genetik tisu penghubung. Pesakit mempunyai simptom yang berlainan dalam tahap yang berbeza: jari panjang dan kaki yang sempit, kerosakan atau saluran darah. Tidak ada ubat untuk sindrom Marfan. Pemeriksaan berkala dapat mencegah komplikasi. Baca semua mengenai Sindrom Marfan di sini!
Kod ICD untuk penyakit ini: Kod ICD adalah kod yang diiktiraf di peringkat antarabangsa untuk diagnosis perubatan. Mereka boleh didapati, misalnya, dalam surat doktor atau sijil ketidakupayaan untuk bekerja. S87
Sindrom Marfan: keterangan
Sindrom Marfan adalah penyakit genetik yang ditularkan dari ibu bapa kepada anak atau berkembang secara spontan. Penyakit yang berkembang secara spontan juga dikenal sebagai penyakit sporadis. Ini berlaku untuk sekitar 25 hingga 30 peratus pesakit dengan sindrom Marfan. Secara keseluruhan, satu hingga lima dari 10.000 orang dalam populasi dipengaruhi oleh sindrom Marfan. Tidak ada perbezaan antara jantina.
Sindrom Marfan: Gejala
Tanda-tanda sindrom Marfan sangat berbeza dan berbeza pada individu pesakit. Walaupun dalam keluarga yang sama, gejala sindrom Marfan dapat sangat berbeza antara anggota keluarga yang sakit. Pelbagai sistem organ dipengaruhi oleh penyakit ini. Yang paling biasa adalah perubahan
- Sistem kardiovaskular
- kerangka
- mata
Sindrom Marfan: Sistem Kardiovaskular
Pesakit dengan sindrom Marfan berisiko meningkat dengan kematian mendadak. Sebabnya adalah air mata yang kerap berlaku di dinding arteri utama (pembedahan aorta). Sebagai hasil dari pembentukan celah dalam dinding aorta, darah tidak lagi diangkut ke saluran darah yang lebih kecil, melainkan meresap ke celah. Risiko pembedahan aorta meningkat pada pesakit sindrom Marfan kerana aorta mereka, yang mempunyai dinding yang lemah, semakin melebar (dilatasi aorta progresif).
Di samping itu, pesakit sering mengalami kerosakan injap jantung seperti regurgitasi aorta dan mitral. Ini boleh menyebabkan aritmia jantung. Selanjutnya, mereka berisiko mengalami radang jantung (endokarditis) dan kegagalan jantung.
Sindrom Marfan: Kerangka
Perubahan rangka selalunya merupakan tanda pertama sindrom Marfan. Pesakit menonjol kerana perawakannya yang tinggi dan kaki yang sangat sempit dan panjang. Jari labah-labah (arachnodactyly) adalah gejala yang terkenal. Jari labah-labah disebut sedemikian kerana sangat panjang dan sempit.
Di samping itu, banyak pesakit mengalami kecacatan dada seperti dada ayam atau corong. Seiring perubahan rangka, mereka sering menderita scoliosis, bengkok dan pusing tulang belakang. Di samping itu, beberapa pesakit mempunyai tulang muka yang kurang berkembang, seperti tulang pipi atau rahang atas.
Keseluruhan perubahan rangka ini juga dikenal sebagai marfanoid habitus.
Sindrom Marfan: mata
Perubahan pada mata yang disebabkan oleh sindrom Marfan mempengaruhi lensa. Ia sering dialihkan (ectopy lensa). Ini mengancam pesakit dengan kebutaan. Faktor risiko lain untuk rabun adalah rabun. Ia disebabkan oleh bola mata yang terlalu panjang. Perubahan ini juga boleh menyebabkan detasmen retina.
Sindrom Marfan: gejala yang mempengaruhi organ lain
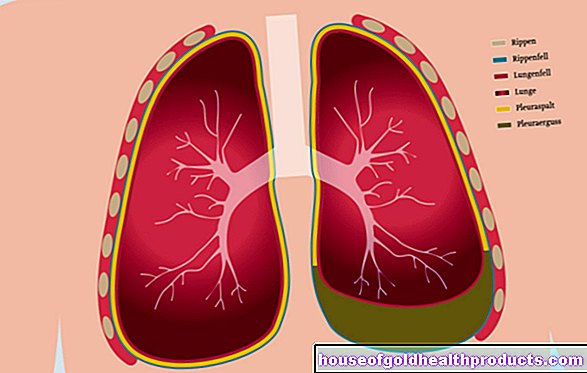
Selain sistem organ yang disebutkan, sindrom Marfan juga dapat merosakkan struktur lain. Ini termasuk paru-paru, antara lain. Mereka yang terjejas mempunyai risiko peningkatan pneumotoraks. Doktor memahami ini bermaksud pengasingan pleura paru-paru dari pleura dan penembusan udara ke celah ini. Keadaan ini boleh mengancam nyawa apabila paru-paru runtuh di kawasan yang terjejas.
Stretch mark sering dilihat pada kulit pesakit dengan sindrom Marfan sebagai tanda tisu penghubung yang lemah.
Dalam perjalanan hidup, apa yang disebut duraektasia dapat berkembang. Ini adalah lanjutan dari meninges, biasanya pada tahap tulang belakang lumbar. Selalunya tidak simptomatik. Dalam beberapa kes, ia boleh menyebabkan kesakitan ketika kantung dural menekan pada saraf tunjang yang keluar.
Sindrom Marfan: Punca dan Faktor Risiko
Sindrom Marfan adalah penyakit keturunan dominan autosomal. Ini bermaksud bahawa ada perubahan (mutasi) pada gen dalam susunan genetik kita yang mencetuskan penyakit. Autosomal dominan menerangkan bahawa maklumat genetik ini terletak pada kompleks gen bukan jantina (autosomal) dan selalu muncul (dominan).
Apabila pesakit dengan sindrom Marfan mempunyai anak, dia boleh mewarisi gen yang berpenyakit atau yang sihat. Kerana setiap orang mempunyai dua set solek genetik. Ini bermaksud bahawa kebarangkalian penghantaran adalah 50 peratus. Seorang kanak-kanak pesakit sindrom Marfan mempunyai 50 peratus kemungkinan penyakit ini.
Sindrom Marfan: tisu penghubung yang rosak
Mutasi yang menyebabkan sindrom Marfan terdapat pada lengan panjang kromosom 15 (15q21). Ia mempengaruhi gen FBN1 yang disebut. Gen ini bertanggungjawab untuk pembentukan protein tisu penghubung, fibrillin-1. Fibrillin-1 penting untuk kestabilan tisu penghubung. Sekiranya pembentukannya dihalang oleh mutasi, tisu penghubung akan kehilangan kestabilan.
Sindrom Marfan: pelbagai bentuk
Keterukan sindrom Marfan berbeza-beza. Doktor kemudian bercakap mengenai ekspresiviti berubah-ubah. Ini bermaksud bahawa gejala pesakit juga berbeza dalam sebuah keluarga. Walaupun terdapat mutasi yang sama, pesakit hampir tidak mempunyai gejala, sementara saudara menunjukkan gambaran lengkap mengenai sindrom Marfan.
Sindrom Marfan: Penyiasatan dan Diagnosis
Diagnosis sindrom Marfan sering dibuat oleh pakar pediatrik. Secara keseluruhan, pelbagai pakar berperanan dalam diagnosis, rawatan dan nasihat. Sebagai tambahan kepada pakar pediatrik, ini termasuk ahli genetik manusia, pakar kardiologi, ortopedis dan pakar oftalmologi. Sebelum diagnosis akhirnya dibuat, doktor anda akan terlebih dahulu bertanya secara terperinci mengenai sejarah perubatan anda (anamnesis). Dia akan menanyakan soalan berikut yang mungkin antara lain:
- Adakah ahli keluarga menghidapi Sindrom Marfan?
- Adakah anda kadang-kadang merasakan jantung berlumba?
- Adakah anda selalu lebih tinggi daripada yang lain semasa anda kecil?
- Adakah anda rabun jauh?
Ujian Fizikal Sindrom Marfan
Doktor anda kemudian akan melakukan pemeriksaan fizikal. Dengan berbuat demikian, dia pertama kali melihat kerangka tersebut. Dia memperhatikan panjang tulang individu, bentuk dada dan bentuk wajah. Kemudian dia mendengar jantung dan paru-paru. Aritmia jantung atau bunyi aliran dapat dilihat di atas arteri utama.
Untuk membuat diagnosis sindrom Marfan, kriteria Gent yang disebut telah dikembangkan. Ia menyenaraikan pelbagai gejala penyakit dalam pelbagai bentuk. Apabila sejumlah kriteria terpenuhi, diagnosis dapat dibuat.
Ujian sindrom Marfan genetik juga mungkin dilakukan. Pembentukan genetik dianalisis dan mutasi yang bertanggungjawab terhadap penyakit itu dicari. Sekiranya terdapat kes sindrom Marfan dalam keluarga, diagnosis yang tepat dapat dibuat sebelum kelahiran.
Sindrom Marfan: Gambar klinikal yang serupa
Penyakit genetik lain yang boleh menyebabkan gejala serupa mesti dibezakan dari sindrom Marfan. Ini termasuk, antara lain
- Sindrom Ehlers-Danlos
- Sindrom Loeys-Dietz
- Sindrom Sphrintzen-Goldberg
- Sindrom MASS
Sindrom Marfan: Rawatan
Oleh kerana sindrom Marfan adalah penyakit genetik, penyebabnya, mutasi, tidak dapat diatasi. Tujuan terapi ini adalah pemeriksaan pesakit secara berkala oleh pelbagai pakar untuk mencegah komplikasi. Yang paling penting, pemantauan jantung adalah penting untuk mengelakkan kematian secara tiba-tiba akibat pembedahan aorta. Ini dapat dicapai dengan mengatasi pelebaran aorta dengan memberikan beta blocker dan mengehadkan aktiviti fizikal. Pelebaran aorta dapat dipantau dengan pemeriksaan ultrasound tahunan dan akar aorta dapat diperbaiki pada masa yang tepat sebelum pembedahan.
Operasi lebih lanjut yang mungkin diperlukan dalam kes sindrom Marfan
- Pembetulan Scoliosis
- Pembetulan dada
- Pembuangan lensa
Sindrom Marfan: kursus penyakit dan prognosis
Kebarangkalian mutasi ditularkan dari satu ibu bapa kepada anak adalah 50 peratus. Pasangan dengan pasangan dengan sindrom Marfan yang merancang untuk mempunyai anak harus mendapatkan nasihat daripada ahli genetik manusia.
Hari ini, jangka hayat dan kualiti hidup hampir tidak terhad untuk pesakit dengan sindrom Marfan. Walaupun jangka hayat jelas terhad pada masa lalu, ia telah meningkat sebanyak 30 tahun berbanding 30 tahun yang lalu. Walau bagaimanapun, pesakit masih berisiko tinggi melakukan pembedahan aorta, yang boleh mengakibatkan kematian secara tiba-tiba. Pembedahan aorta paling kerap dilihat pada usia 30 tahun. Pemeriksaan berkala oleh pakar rawatan dapat mengurangkan risiko pembedahan aorta pada sindrom Marfan.
Tags.: temu ramah simptom parasit









.jpg)