hemofilia
dan Martina Feichter, penyunting perubatan dan ahli biologiDr. med. Fabian Sinowatz adalah freelancer dalam pasukan editorial perubatan
Lebih banyak mengenai pakarMartina Feichter belajar biologi dengan farmasi subjek elektif di Innsbruck dan juga melibatkan diri dalam dunia tanaman perubatan. Dari situ tidak jauh dari topik perubatan lain yang masih memikatnya hingga ke hari ini. Dia dilatih sebagai wartawan di Axel Springer Academy di Hamburg dan telah bekerja di sejak 2007 - pertama sebagai penyunting dan sejak 2012 sebagai penulis bebas.
Lebih banyak mengenai pakar Semua kandungan diperiksa oleh wartawan perubatan.
Haemophilia (penyakit darah) adalah gangguan pembekuan darah yang biasanya diwarisi. Penderita kekurangan faktor pembekuan darah yang penting atau mereka cacat. Akibatnya, hemofilia terdedah kepada pendarahan dan lebam dengan mudah. Hemofilia belum dapat disembuhkan. Oleh kerana terapi moden, hemofilia dapat menjalani kehidupan yang normal. Baca semua yang anda perlu ketahui mengenai hemofilia di sini.
Kod ICD untuk penyakit ini: Kod ICD adalah kod yang diiktiraf di peringkat antarabangsa untuk diagnosis perubatan. Mereka boleh didapati, misalnya, dalam surat doktor atau sijil ketidakupayaan untuk bekerja. D66D67D68
Gambaran ringkas
- Apa itu Hemofilia? Gangguan pembekuan darah genetik (koagulopati). Ia juga disebut hemofilia.
- Bentuk hemofilia: Yang paling biasa adalah hemofilia A, diikuti oleh hemofilia B. Bentuk lain seperti haemophilia C, sindrom von Willebrand-Jürgens dan parahemophilia kurang biasa.
- Sebab: Kekurangan atau kekurangan faktor pembekuan (protein darah yang diperlukan untuk darah membeku). Kebanyakannya diwarisi dan jarang diperoleh (disebabkan oleh mutasi gen spontan).
- Gejala: peningkatan kecenderungan untuk berdarah, yang boleh menyebabkan pendarahan dan lebam dengan mudah (hematoma). Pendarahan juga memakan masa lebih lama daripada biasa. Seberapa teruk gejala bergantung pada keparahan hemofilia.
- Diagnostik: Pengukuran pelbagai parameter darah (aPTT, Nilai cepat, masa trombin plasma, masa pendarahan, bilangan platelet darah), penentuan aktiviti faktor pembekuan
- Rawatan: penggantian faktor pembekuan yang hilang (dalam bentuk pekatan faktor); dalam kes tertentu ubat lain (seperti desmopressin untuk hemofilia A ringan)
Hemofilia: keterangan
Hemofilia adalah gangguan kongenital pembekuan darah. Mereka yang terkena (hemofilia, "hemofiliacs") tidak dapat membentuk faktor pembekuan yang cukup berfungsi. Ini adalah protein dalam darah yang diperlukan agar darah membeku. Kerana kekurangan faktor pembekuan, gumpalan darah dan luka tidak dapat terbentuk dengan mudah. Oleh itu, pesakit hemofilia terdedah kepada pendarahan. Mereka juga mengalami luka yang berdarah lebih lama daripada biasa. Ini boleh membahayakan dalam keadaan tertentu.
Istilah perubatan untuk gangguan pembekuan darah dengan peningkatan kecenderungan pendarahan adalah "diatesis hemoragik". Nama umum untuk gangguan pembekuan adalah "koagulopati".
Hemofilia: kekerapan
Hemofilia berlaku hampir secara eksklusif pada kanak-kanak lelaki dan lelaki. Ia jarang berlaku: hanya sekitar dua dari 10,000 lelaki yang menderita hemofilia. Terdapat sekitar 10,000 orang yang terjejas di Jerman. Kira-kira 3,000 hingga 5,000 dari mereka mempunyai bentuk penyakit yang teruk.
Hemofilia: bentuk
Terdapat faktor pembekuan yang berbeza. Bergantung pada faktor pembekuan yang mempengaruhi hemofilia, profesional perubatan membezakan antara pelbagai bentuk penyakit.
Hemofilia A.
Pada hemofilia A terdapat masalah dengan faktor pembekuan VIII (antihemofilik globulin A): Sama ada badan tidak dapat menghasilkannya dalam jumlah yang mencukupi atau rosak. Kira-kira 85 peratus daripada semua hemofilia menderita hemofilia A. Hampir kesemuanya adalah lelaki.
Hemofilia B.
Pada hemofilia B, faktor pembekuan IX (antihemophilic globulin B atau Christmas factor) hilang. Di sini juga, pesakit kebanyakannya lelaki. Pada masa lalu, hemofilia B berlaku lebih kerap pada keluarga kerajaan Inggeris dan keluarga tsaris Rusia. Itulah sebabnya ia juga disebut "penyakit raja".
Bentuk hemofilia lain
Sebagai tambahan kepada bentuk utama hemofilia A dan B, terdapat gangguan pembekuan keturunan yang lain. Salah satunya ialah sindrom von Willebrand-Jürgens, juga disebut sindrom von Willebrand (vWS). Fokus di sini adalah pada faktor von Willebrand: faktor pembekuan ini sama ada terlalu kecil atau rosak. Seperti hemofilia A dan B, ini menyebabkan peningkatan kecenderungan pendarahan (diatesis hemoragik). Di Jerman, sindrom von Willebrand-Jürgens dijumpai pada sehingga satu peratus populasi. Berbeza dengan dua bentuk utama hemofilia, gangguan pembekuan ini mempengaruhi lelaki dan wanita secara sama.
Dalam kes yang jarang berlaku, gangguan pembekuan lain juga terjadi, yang berdasarkan kekurangan faktor pembekuan. Mereka juga mengalami kecenderungan pendarahan yang meningkat (diatesis hemoragik). Ini termasuk:
- Haemophilia C: Kekurangan faktor fungsi XI
- Parahemophilia: kekurangan faktor fungsi V
- Hipoprokonvertinemia: Kekurangan faktor fungsi VII
- Kekurangan Faktor Stuart Prower: Kekurangan faktor kerja X
- Sindrom Hageman: Kekurangan Fungsi Berfungsi XII
- Kekurangan fibrinase: kekurangan faktor fungsi XIII
Hemofilia: gejala
Gejala hemofilia A (kekurangan faktor VIII) dan hemofilia B (kekurangan faktor IX) adalah sama - walaupun terdapat faktor pembekuan yang berbeza dalam setiap kes. Secara umum, seseorang boleh mengatakan: semakin sedikit faktor pembekuan berfungsi, semakin jelas gambaran klinikalnya.
Hemofilia: tahap keparahan
Keterukan hemofilia bergantung pada seberapa besar aktiviti faktor pembekuan dikurangkan berbanding fungsinya pada orang yang sihat. Sekiranya aktiviti faktor hanya sedikit berkurang, mereka yang terjejas biasanya tidak mempunyai gejala apa pun. Sebaliknya, dalam haemophiles simptomatik, aktiviti faktor lebih berkurang, yang menyebabkan tanda-tanda yang lebih jelas. Terdapat tiga tahap keparahan hemofilia (A dan B):
Hemofilia ringan:Faktor aktiviti adalah 6 hingga 45 peratus daripada aktiviti normal orang yang sihat; sekitar 20 peratus daripada semua penderita hemofilia mempunyai tahap keparahan ini. Mereka yang terjejas sering tidak menyedari banyak penyakit mereka. Inilah sebabnya mengapa hemofilia hanya ditemui pada banyak orang pada masa remaja atau dewasa, ketika operasi atau kecederaan besar berdarah lebih lama dari yang dijangkakan.
Hemofilia sederhana: Faktor aktiviti adalah 1 hingga 5 peratus aktiviti normal; kira-kira 20 peratus daripada semua hemofil juga terjejas. Gejala biasanya menjadi jelas pada beberapa tahun pertama kehidupan dengan pendarahan yang luar biasa lama dan lebam yang kerap. Seperti hemofilia ringan, pendarahan biasanya disebabkan oleh kecederaan atau pembedahan. Pendarahan spontan jarang berlaku.
Hemofilia yang teruk: Faktor aktiviti kurang daripada 1 peratus aktiviti normal. Ini berlaku untuk sekitar 55 peratus dari semua hemofilia. Hemofilia biasanya sudah terlihat ketika dilahirkan: membuang tali pusat memicu pendarahan besar-besaran. Mimisan berat tidak jarang berlaku pada masa bayi. Selain itu, kecederaan atau lebam terkecil boleh menyebabkan pendarahan yang teruk di bawah kulit (lebam). Pendarahan dalaman juga lebih biasa, contohnya pendarahan yang menyakitkan ke sendi besar (seperti lutut, siku). Biasanya, banyak pendarahan tidak mempunyai sebab yang jelas (pendarahan spontan).
Hemofilia: Risiko dan Bahaya
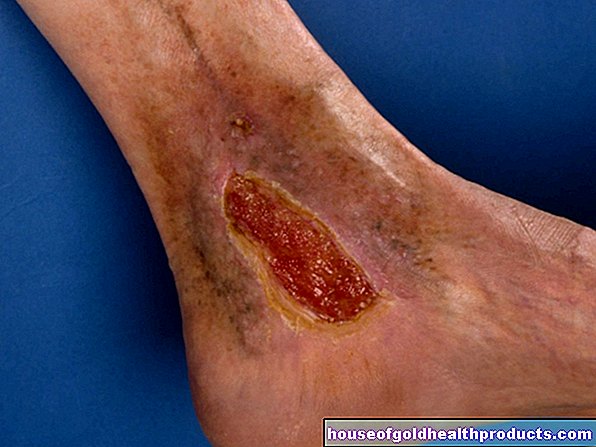
Pendarahan ke sendi (hemarthrosis) sering berlaku berulang kali, terutama pada hemofilia yang teruk. Akibatnya, sendi yang terjejas boleh cacat, haus sebelum waktunya (osteoartritis) dan secara beransur-ansur kaku. Orang yang menderita hemarthosis hampir tidak dapat menggerakkan lengan dan kaki mereka dan bergantung pada kerusi roda.
Bahaya lain dengan hemofilia adalah pendarahan ke otot: ia boleh merosakkan tisu otot dan menyebabkan kelemahan otot.
Pada prinsipnya, pendarahan dalaman pada hemofilia dapat terjadi di mana-mana kawasan organ. Pendarahan di otak jarang berlaku tetapi berbahaya: sebagai contoh, ia boleh merosakkan kemampuan berfikir dan mengurangkan tumpuan. Pendarahan serebrum yang teruk boleh membawa maut! Pendarahan di perut juga boleh mengancam nyawa dalam keadaan tertentu. Pendarahan berat di orofaring boleh mempengaruhi saluran udara.
Selain kecenderungan pendarahan (diatesis hemoragik), hemofilia dari tahap keparahan juga dapat menyebabkan gangguan penyembuhan luka.
Sindrom Von Willebrand-Juergens: Gejala
Orang dengan sindrom von Willebrand-Jürgens juga mempunyai peningkatan kecenderungan untuk berdarah. Selalunya, terdapat pendarahan ringan di bawah kulit (lebam), gusi berdarah atau pendarahan yang berpanjangan setelah pembedahan, misalnya setelah pencabutan gigi. Tetapi ada juga pesakit yang mengalami pendarahan berat.
Hemofilia: rawatan
Terapi hemofilia bergantung pada jenis dan keparahan hemofilia. Doktor yang hadir akan membuat rancangan terapi yang sesuai untuk setiap pesakit. Selain rawatan ubat, dia juga dapat mengesyorkan langkah-langkah umum. Sebagai contoh, sangat disarankan, terutama dalam kes hemofilia yang teruk, untuk melakukan rawatan fizikal dan mengelakkan sukan tertentu (berbahaya).
Hemofilia A dan B
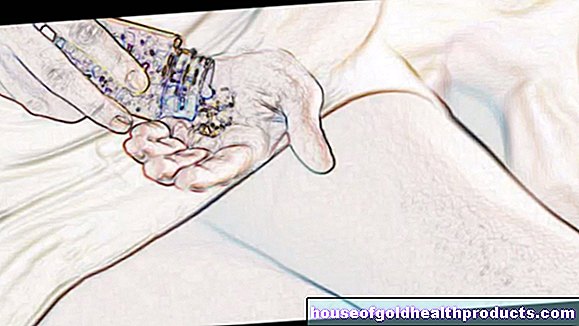
Hari ini, pekatan faktor yang disebut tersedia untuk rawatan hemofilia. Ini adalah konsentrat faktor pembekuan darah VIII (untuk hemofilia A) atau IX (untuk hemofilia B) yang diperoleh dari plasma darah atau direkayasa secara genetik. Mereka harus disuntik ke vena (secara intravena). Ramai penghidap belajar untuk menyuntikkan faktor tersebut. Ini memberi mereka banyak kebebasan dalam menangani penyakit ini.
Pada tahun 1960-an dan 1970-an, banyak orang di Jerman dijangkiti hepatitis dan / atau HIV dari persiapan faktor yang tercemar. Ini secara praktikalnya tidak lagi berlaku hari ini: plasma darah dikawal dengan ketat dan dirawat sebelum ia diberikan. Dengan kepekatan faktor rekayasa genetik secara amnya tidak ada risiko jangkitan.
Sekiranya hemofilia ringan dan sederhana, pemberian konsentrat faktor hanya diperlukan jika perlu (rawatan berdasarkan permintaan): Antikoagulan diberikan, misalnya, sekiranya berlaku pendarahan yang lebih tinggi atau sebelum operasi yang dirancang. Kecederaan ringan seperti lecet tidak perlu dirawat dengan faktor pekat. Pendarahan biasanya dapat dihentikan dengan memberi tekanan ringan ke kawasan pendarahan.
Orang yang menderita hemofilia yang teruk, sebaliknya, perlu menyuntik diri dengan faktor pekat secara berkala (rawatan jangka panjang). Faktor VIII dalam hemofilia A diberikan dua hingga tiga kali seminggu. Faktor IX pada hemofilia B umumnya hanya perlu disuntik sekali atau dua kali seminggu kerana masa penahanannya lebih lama dalam darah.
Pembedahan dan kecederaan akut
Sebelum operasi (walaupun pengambilan gigi dirancang), terapi persediaan mesti dijalankan untuk semua pesakit hemofilia. Kepekatan faktor biasanya diberikan untuk tujuan ini. Ini adalah satu-satunya cara untuk mencegah komplikasi serius daripada kehilangan darah yang berlebihan semasa pembedahan.
Dalam kes hemofilia A ringan, bukannya faktor pekat, ubat yang menstabilkan pembekuan darah dapat diberikan sebelum operasi yang dirancang. Ini termasuk desmopressin, sebagai contoh. Ini adalah protein yang dihasilkan secara buatan. Ia merangsang pembebasan faktor VIII yang tersimpan dari saluran darah. Ubat itu hanya boleh digunakan selama beberapa hari, jika tidak, kedai-kedai akan segera kosong.
Petua: Sebelum prosedur yang dirancang, penderita hemofilia harus berbincang dengan ahli haematologi atau pusat hemofilia mereka yang mana terapi pencegahan disarankan dalam kes mereka.
Sekiranya berlaku kecederaan akut semasa kecemasan, selain langkah hemostasis tempatan (contohnya pembalut tekanan), perlu dilakukan pemberian pekatan faktor.
Pekat faktor: komplikasi
Sebilangan orang membentuk antibodi (perencat) terhadap faktor pembekuan dalam faktor pekat. Hemofilia penghambat yang disebut ini lebih kerap berlaku pada orang dengan hemofilia A daripada pada orang yang menderita hemofilia B. Perencat tidak mematikan faktor pembekuan tambahan. Terapi itu tidak seefektif yang diinginkan. Haemophilia B juga mengancam reaksi alahan yang teruk dan komplikasi lain.
Jumlah perencat dalam darah diberikan dalam unit Bethesda (BE) yang disebut. Semakin tinggi nilai BE, semakin banyak perencat yang terdapat dalam darah pesakit.
Pada hemofilia A, penumpukan sedikit perencat sering dapat dikompensasi dengan meningkatkan dos pekatan faktor. Sekiranya perencat terbentuk dalam jumlah besar, terapi toleransi imun disyorkan: pesakit menerima dos yang sangat tinggi dari faktor pembekuan yang hilang dalam rejimen terapi yang kompleks. Sistem imun secara beransur-ansur terbiasa dengan kehadirannya dan menghentikan pembentukan perencat.
Hemofilia inhibitor yang jarang berlaku pada hemofilia B diperlakukan secara berbeza. Contohnya, mereka yang terjejas menerima ubat yang mempengaruhi sistem imun (imunomodulator).
Ubat tahan sakit
Hemofilia yang serius boleh menyebabkan kesakitan yang teruk bagi mereka yang terjejas. Contohnya, pendarahan pada sendi boleh menjadi sangat menyakitkan. Kemudian penghilang rasa sakit seperti ibuprofen membantu. Sebaliknya, asid acetylsalicylic penghilang rasa sakit (ASA) tidak sesuai untuk hemofilia: ia meningkatkan kecenderungan untuk berdarah lebih jauh (kesan sampingan ASA).
Sindrom von Willebrand-Juergens (VWS)
Dalam sindrom von Willebrand-Jürgens, perbezaan dibuat antara pelbagai jenis, yang diperlakukan secara berbeza: Dalam jenis 1 yang disebut, bahan aktif desmopressin diberikan apabila perlu (sebelum operasi atau sekiranya berlaku pendarahan akut). Ia merangsang pembebasan faktor pembekuan yang tersimpan.
Desmopressin juga digunakan dalam jenis 2; bagaimanapun, ubat itu tidak selalu berfungsi di sini. Kemudian doktor menetapkan faktor pekat (dengan faktor von Willebrand) sebagai gantinya.
Pesakit dengan VWS jenis 3 selalu dirawat dengan faktor pekat.
Hemofilia: penyebab dan faktor risiko
Hemofilia adalah kelainan genetik kongenital yang biasanya diwarisi. Ia berlaku lebih jarang secara spontan (akibat perubahan gen spontan = mutasi spontan).
Pada hemofilia, maklumat genetik yang diperlukan untuk menghasilkan faktor pembekuan berfungsi tidak betul: pada hemofilia A, itu adalah faktor pembekuan VIII, pada hemofilia B, faktor IX. Hasil rancangan pembinaan yang salah adalah faktor pembekuan yang relevan tidak dapat dihasilkan dalam kuantiti yang cukup berfungsi. Ini mengganggu pembekuan darah: luka tidak menutup dengan cepat, sehingga pendarahan memerlukan waktu yang sangat lama. Beberapa pesakit juga mengalami pendarahan spontan (tanpa sebab yang jelas).
Hemofilia A dan B: pewarisan
Cetakan biru (gen) untuk semua bahagian badan terdapat pada kromosom. Dalam nukleus setiap sel dalam tubuh terdapat 46 kromosom, termasuk dua kromosom seks, yang antara lain menentukan jantina. Wanita mempunyai dua kromosom seks X (XX): Satu kromosom X masing-masing diwarisi dari ibu dan satu dari bapa. Lelaki, sebaliknya, mempunyai kromosom Y yang diwarisi dari bapa dan kromosom X diwarisi dari ibu (XY).
Gen untuk faktor pembekuan berada pada kromosom X. Pada wanita yang mempunyai dua kromosom X, jika salah satunya mengandung cetak biru yang salah untuk faktor pembekuan, ini biasanya dapat dikompensasi oleh kromosom X yang lain. Oleh itu, mereka kebanyakannya bebas gejala sepanjang hayat mereka.
Sekiranya wanita seperti itu mempunyai anak, dia menyebarkan salah satu daripada dua kromosom X ke keturunan. Kebarangkalian bahawa ini adalah salinan dengan cetak biru yang salah adalah 50 peratus. Dengan meneruskan kecacatan genetik, ibu menjadi pembawa (pembawa) hemofilia. Sekiranya ada anak lelaki, dia akan dilahirkan sebagai hemofilia. Sebaliknya, anak perempuan biasanya menjadi bakal pembawa.
Dalam kes yang jarang berlaku, faktor pembekuan yang relevan tidak cukup terbentuk walaupun pada konduktor wanita. Luka akibat kecederaan atau operasi kemudiannya dapat berdarah dalam jangka masa yang lama. Lebih jarang bagi seorang gadis untuk mewarisi kromosom X yang rosak dari kedua ibu bapa. Ini kebanyakan berkaitan dengan sindrom Turner penyakit keturunan. Gadis-gadis yang terkena menunjukkan gambaran penuh mengenai hemofilia.
Sindrom Von Willebrand-Juergens
Dalam sindrom von Willebrand-Jürgens, petunjuk untuk faktor von Willebrand (vWF) menunjukkan mutasi: faktor pembekuan terlalu kecil atau rosak. Mutasi gen boleh berlaku pada lelaki dan wanita.
Hemofilia: pemeriksaan dan diagnosis
Sekiranya seseorang sering mengalami pendarahan spontan (seperti mimisan) atau lebam dengan mudah, ini boleh menjadi tanda hemofilia. Kecurigaan ini sangat jelas jika keluarga telah mengetahui kes-kes hemofilia. Mereka yang terjejas harus mempunyai kecenderungan peningkatan pendarahan yang diperjelaskan oleh doktor. Titik hubungan pertama jika anda mengesyaki hemofilia adalah doktor keluarga anda:
Doktor akan terlebih dahulu mengambil riwayat perubatan pesakit (anamnesis) dalam perbincangan dengan pesakit: Dia mempunyai gejala yang dijelaskan secara terperinci, bertanya tentang sebarang penyakit yang mendasari dan adakah terdapat kes hemofilia yang diketahui dalam keluarga.
Ujian makmal sangat penting untuk menjelaskan kemungkinan hemofilia. Doktor mengambil sampel darah dari pesakit untuk memeriksanya di makmal untuk mengetahui beberapa parameter: Dalam hemofilia, aPTT (masa tromboplastin separa diaktifkan) lebih lama daripada pada orang yang sihat. Sebaliknya, nilai Pantas yang disebut (masa tromboplastin, TPZ) dan masa trombin plasma (PTZ) umumnya normal; mereka hanya berpanjangan dalam hemofilia yang teruk. Bilangan platelet darah (trombosit) dan masa pendarahan yang disebut juga normal pada hemofilia A dan B. Dalam sindrom von Willebrand-Jürgens, sebaliknya, masa pendarahan berpanjangan. Waktu pendarahan adalah jumlah masa yang diperlukan untuk menghentikan pendarahan.
Untuk dapat menentukan hemofilia A atau B dengan jelas, aktiviti faktor pembekuan (VIII, IX) mesti dianalisis. Pakar dalam bidang hematologi atau pusat perubatan khusus (pusat hemofilia) bertanggungjawab untuk ini.
Hemofilia: ujian pada bayi baru lahir dan bayi yang belum lahir
Sekiranya keluarga telah menderita hemofilia, pembekuan bayi baru lahir lelaki biasanya diperiksa sebaik sahaja dilahirkan. Dengan cara ini, hemofilia dapat dikesan pada peringkat awal. Anda juga boleh memeriksa hemofilia semasa mengandung.
Sekiranya seorang wanita mengesyaki bahawa dia mempunyai kecenderungan genetik terhadap hemofilia dan oleh itu merupakan pembawa berpotensi, ini dapat dijelaskan dengan ujian genetik.
Hemofilia: kursus penyakit dan prognosis
Hemofilia belum disembuhkan. Pesakit harus mengatasi kekurangan faktor pembekuan sepanjang hidup mereka. Dengan bantuan faktor pekat, mereka biasanya dapat menjalani kehidupan yang normal.
Sekiranya tidak dirawat, hemofilia sederhana dan teruk sering menyebabkan komplikasi serius. Contohnya, pendarahan ke otot boleh menyebabkan kerosakan otot. Pendarahan pada sendi boleh menyebabkan osteoartritis dan kekejangan sendi. Komplikasi seperti itu dapat dielakkan jika hemofilia dikesan lebih awal dan dirawat secara konsisten.
Tags.: terapi anatomi temu ramah