Atrofi otot tulang belakang
dan Florian Tiefenböck, doktor Dikemas kini padaMaximilian Reindl belajar kimia dan biokimia di LMU di Munich dan telah menjadi ahli pasukan editorial sejak Disember 2020. Dia akan membiasakan diri dengan topik perubatan, saintifik dan kesihatan untuk anda agar mereka dapat difahami dan difahami.
Lagi catatan oleh Maximilian ReindlFlorian Tiefenböck belajar perubatan manusia di LMU Munich. Sejak itu, dia menyertai sebagai pelajar pada bulan Mac 2014 dan telah menyokong pasukan editorial dengan artikel perubatan sejak itu. Setelah mendapat lesen perubatan dan pekerjaan praktikalnya dalam perubatan dalaman di University Hospital Augsburg, dia telah menjadi anggota tetap pasukan sejak Disember 2019 dan, antara lain, memastikan kualiti perubatan alat
Lagi kiriman oleh Florian Tiefenböck Semua kandungan diperiksa oleh wartawan perubatan.
Atrofi otot tulang belakang, atau SMA, adalah penyakit yang jarang berlaku di mana sel-sel saraf tertentu di saraf tunjang mati. Rangsangan dan dorongan dari otak kemudian tidak lagi sampai ke destinasinya: otot. Ini menyebabkan pembaziran otot dan kelumpuhan. Terdapat pelbagai bentuk SMA. Yang paling teruk bermula pada masa bayi. Rawatan baru menjanjikan peningkatan kesihatan yang berkekalan. Baca lebih lanjut mengenai atrofi otot tulang belakang di sini.
Kod ICD untuk penyakit ini: Kod ICD adalah kod yang diiktiraf di peringkat antarabangsa untuk diagnosis perubatan. Mereka boleh didapati, misalnya, dalam surat doktor atau sijil ketidakupayaan untuk bekerja. G12

Gambaran ringkas
- Apakah atrofi otot tulang belakang? Sekumpulan penyakit kelemahan otot. Mereka didasarkan pada kematian sel-sel saraf tertentu di saraf tunjang yang mengawal otot (neuron motorik). Oleh itu, SMA adalah antara penyakit neuron motor.
- Apa bentuk yang ada? Atrofi otot tulang belakang keturunan kebanyakannya SMA dengan kecacatan genetik tertentu pada kromosom 5 (SMA yang berkaitan dengan 5q). Doktor membezakan antara empat bentuk yang berbeza: SMA jenis 1 - jenis 4. Di samping itu, terdapat bentuk sporadis, keturunan yang tidak pasti.
- Kekerapan: Penyakit Jarang; SMA yang diwarisi mempengaruhi kira-kira satu bayi baru lahir pada tahun 7000.
- Gejala: kekejangan otot, kelemahan otot progresif, pembaziran otot, kelumpuhan. Kecerunan berbeza bergantung pada bentuk SMA.
- Punca: Jenis atrofi otot tulang belakang keturunan 1-4 adalah hasil daripada kecacatan genetik pada kromosom 5, lebih tepat pada gen SMN1. Akibatnya, tubuh kekurangan protein khas, protein SMN. Kekurangan ini merosakkan neuron motor pada saraf tunjang.
- Diagnosis: Pemeriksaan genetik untuk susunan genetik SMN yang diubah, pemeriksaan fizikal, elektroneurografi, elektromiografi, ujian darah (mis. CK)
- Rawatan: Terapi penggantian gen atau pemberian ubat penyekat penyambungan adalah mungkin. Menemani fisioterapi, terapi pertuturan, terapi kesakitan dan psikoterapi. Sekiranya perlu, pembedahan pada tulang belakang. Pelan rawatan bergantung pada jenis SMA.
- Prognosis: Dalam hal SMA keturunan keturunan, pilihan terapi baru mempunyai kesan kausal dan boleh memberi kesan positif terhadap perjalanan penyakit ini. Memulakan rawatan lebih awal adalah mustahak. Rawatan belum tersedia untuk setiap pesakit. Sekiranya tidak dirawat, kanak-kanak dengan SMA jenis 1 biasanya mati dalam dua tahun pertama. Jangka hayat dalam jenis 3 dan jenis 4 hampir atau tidak berkurang.
Apakah atrofi otot tulang belakang?
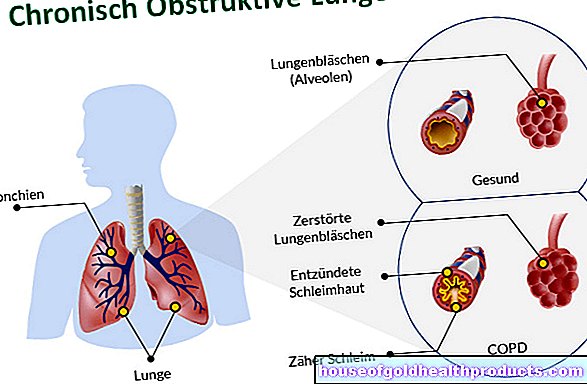
Dalam atrofi otot tulang belakang (SMA), sel saraf tertentu di saraf tunjang mati. Mereka biasanya mengawal otot, sebab itulah para pakar menyebut sel-sel saraf ini sebagai neuron motorik. Oleh itu, SMA tergolong dalam penyakit neuron motor yang disebut.
Dalam kes atrofi otot tulang belakang, neuron motor bawah (kedua), yang secara langsung dihubungkan ke otot dengan pelengkap mereka, terjejas. Akibat kerosakan itu, sedikit atau lebih banyak isyarat saraf sampai ke otot. Otot menjadi semakin lemah dan semakin kecil (otot membuang / atrofi otot).
Doktor membezakan antara pelbagai bentuk atrofi otot tulang belakang. Sejauh ini kumpulan terbesar adalah SMA keturunan, di mana otot yang dekat dengan batang (proksimal) terjejas. Mereka berdasarkan kecacatan genetik tertentu. Kira-kira satu daripada 7,000 bayi baru lahir akan mengembangkannya.
Atrofi otot tulang belakang adalah penyakit yang jarang berlaku secara keseluruhan. Walaupun begitu, ia adalah penyakit yang diwarisi secara resesif autosomal kedua yang paling biasa. Ia juga merupakan penyebab kematian yang paling biasa pada bayi atau kanak-kanak kerana kecacatan genetik.
Apa jenis atrofi otot tulang belakang?
Doktor membezakan bentuk keturunan (keturunan) SMA daripada bentuk sporadis. Satu lagi klasifikasi atrofi otot tulang belakang terutama berkaitan dengan kumpulan otot yang terjejas terlebih dahulu. Disana ada
- SMA Proksimal: Dengan sekitar 90 peratus, mereka membentuk kumpulan SMA terbesar. Gejala bermula pada otot berhampiran batang badan, iaitu berdekatan.
- SMA bukan proksimal: Di sini, kumpulan otot yang lebih jauh, seperti tangan dan kaki, terjejas terlebih dahulu (SMA distal). Dalam kursus selanjutnya, SMA ini juga boleh merebak ke otot berhampiran bahagian tengah badan.
- Bentuk khas (mis. Atrofi otot spinobulbar jenis Kennedy)
Atrofi otot tulang belakang proksimal
Atrofi otot tulang belakang proksimal keturunan kebanyakannya penyakit yang berdasarkan pada kecacatan genetik tertentu (SMA yang berkaitan dengan 5q, kecacatan pada kromosom 5). Ini seterusnya dibahagikan kepada empat bentuk yang berbeza. Klasifikasi dibuat berdasarkan masa di mana gejala pertama muncul dan perjalanan penyakit.
Atrofi Otot Tunjang Jenis 1: Ini adalah bentuk SMA yang paling biasa dan paling serius. Ia juga disebut "penyakit Werdnig-Hoffmann" atau "SMA infantil akut". Penyakit ini biasanya bermula pada awal bayi. Kelemahan otot mempengaruhi seluruh badan - doktor juga membicarakan "sindrom bayi floppy" (dari bahasa Inggeris floppy = lembap, bayi = bayi, kanak-kanak). Kebanyakan kanak-kanak yang tidak dirawat dengan SMA jenis 1 mati sebelum mereka berumur dua tahun.
Atrofi otot tunjang jenis 2: Bentuk SMA ini juga dikenali sebagai "atrofi otot tulang belakang menengah" atau "SMA infantil kronik". Gejala pertama biasanya muncul sebelum usia 18 bulan. Mereka yang terjejas mempunyai jangka hayat yang kadang-kadang berkurang dengan ketara.
Atrofi otot tulang belakang jenis 3: Ia juga dikenali sebagai "atrofi otot tulang belakang remaja" atau "penyakit Kugelberg-Welander". SMA ini biasanya bermula selepas usia 18 bulan dan sebelum awal dewasa. Kelemahan otot lebih ringan daripada pada jenis 1 atau 2. Mereka yang terkena hanya mempunyai jangka hayat yang sedikit berkurang.
Atrofi otot tulang belakang jenis 4: Ia serupa dengan jenis SMA 3, tetapi hanya muncul pada masa dewasa (biasanya> 30 tahun). Walau bagaimanapun, kelemahan otot kurang ketara dan berkembang lebih perlahan berbanding dengan SMA jenis 3.
Peralihan antara versi yang berbeza adalah lancar. Dalam beberapa kes, ini menjadikan penentuan jelas sukar. Beberapa kecenderungan genetik juga memainkan peranan penting dalam keparahan penyakit yang dimaksudkan.
Atrofi otot tulang belakang yang lain
Sebagai tambahan kepada bentuk proksimal ini, terdapat bentuk lain atrofi otot tulang belakang. Ini termasuk, misalnya, lebih jarang, juga atrofi otot tulang belakang distal keturunan. Dengan mereka, gejala biasanya bermula pada kumpulan otot yang berada jauh dari badan.
Warisan tidak dijamin dalam kes SMA sporadis. Di samping itu, pengumpulan keluarga tidak dapat ditentukan. Dalam kesusasteraan, ini termasuk:
- Jenis Hirayama (SMA distal remaja, penyakit sekitar usia 15 tahun, mempengaruhi otot lengan, biasanya berhenti walaupun tanpa terapi dan bahkan boleh bertambah baik)
- Jenis Vulpian-Bernhard (juga sindrom "lengan flail" dengan permulaan pada tali pinggang bahu, biasanya dari usia 40 tahun)
- Jenis Duchenne-Aran (mula-mula mempengaruhi otot tangan, merebak ke arah badan, biasanya selepas usia 30 tahun)
- Jenis peroneal (sindrom "flail-leg", pertama pada otot kaki bawah)
- Palsu bulbar progresif (gangguan pertuturan dan menelan, mempengaruhi sekitar 20 peratus pesakit dengan sklerosis lateral amyotrophic)
Beberapa bentuk SMA sporadis (sindrom "flail-arm - / - leg", kelumpuhan bulbar progresif) dikira antara varian sklerosis lateral amyotrophic (ALS) dalam kalangan pakar. Artikel ini membincangkan terutamanya atrofi otot tulang belakang proksimal keturunan.
Atrofi otot spinobulbar
Atrofi otot spinobulbar atau bulbospinal (jenis Kennedy, sindrom Kennedy) adalah penyakit yang diwarisi. Selalunya bermula pada usia dewasa hingga pertengahan. Bentuk SMA khas ini diwarisi secara resesif berkait X dan oleh itu hanya mempengaruhi lelaki (kerana lelaki hanya mempunyai satu kromosom X, pada wanita kedua, kromosom X yang sihat mendominasi dan akan mengimbangi kecacatan).
Kelemahan otot pada otot yang berdekatan dengan badan pada kaki dan lengan atau bahu serta otot lidah dan tekak adalah perkara biasa. Akibatnya, mereka yang terkena masalah menghadapi masalah bercakap dan menelan, misalnya. Mereka juga mengadu gegaran, kekejangan otot dan kedutan. Lelaki yang terjejas juga sering mengalami testis dan steril. Di samping itu, kelenjar susu membesar (ginekomastia).
Atrofi otot spinobulbar biasanya perlahan. Jangka hayat hampir tidak terhad.
Bagaimana anda mengenali atrofi otot tulang belakang?
Atrofi otot tulang belakang yang tipikal adalah kelemahan otot progresif hingga kelumpuhan (paresis) dan otot berkedut. Akibat kerosakan saraf, otot tidak lagi menerima impuls elektrik, yang menyebabkannya menyusut dari masa ke masa (atrofi otot). Tanda dan keluhan yang tepat bergantung pada bentuk masing-masing. Bahagian berikut melihat gejala SMA proksimal keturunan.
Gejala Atrofi Otot Tunjang Infantile Jenis 1
Dengan SMA jenis 1, gejala muncul dalam enam bulan pertama kehidupan. Kelemahan otot secara umum berlaku - iaitu, yang mempengaruhi seluruh badan. Di samping itu, ketegangan antara otot berkurang. Doktor bercakap mengenai hipotonia.
Pada bayi baru lahir, kelemahan otot ini pada mulanya menampakkan diri pada postur kaki yang biasa, yang mengingatkan pada katak yang berbaring (postur kaki katak). Kaki dibengkokkan, lutut bersudut ke luar dan kaki bersudut ke dalam. Walaupun mengangkat atau memegang kepala sendiri tidak mungkin dilakukan.
Pada usia lanjut, kanak-kanak dengan SMA jenis 1 tidak boleh duduk atau berjalan sendiri. Ramai kanak-kanak juga tidak dapat bercakap, kerana otot lidah juga boleh terjejas.
Ciri lain atrofi otot tulang belakang jenis 1 adalah bentuk badan atas: otot-otot dada dan punggung tidak berkembang dengan baik. Ini memberikan bahagian atas bentuk seperti loceng (dada loceng). Kerana perkembangan otot di dada dan punggung yang rendah, mereka yang terjejas menggunakan postur bungkuk.
Selalunya terdapat juga kelengkungan tulang belakang yang semakin meningkat (scoliosis). Postur membongkok ke depan dan membongkok menyebabkan masalah pernafasan lebih jauh. Pernafasan yang sangat cepat dan cetek (tachypnea) adalah ciri.
Gejala atrofi otot tulang belakang pertengahan jenis 2
Atrofi otot tulang belakang jenis 2 biasanya hanya menyebabkan simptom antara usia tujuh hingga 18 bulan. Kanak-kanak yang terjejas boleh duduk sendiri, tetapi biasanya tidak belajar berdiri atau berjalan. Kelemahan otot berkembang lebih perlahan daripada pada jenis 1.
Dengan SMA jenis 2, gejala yang serupa dengan bentuk bayi yang teruk, seperti ubah bentuk tulang belakang, timbul dari masa ke masa. Sendi kaku kerana otot dan tendon yang dipendekkan (kontraksi). Tanda-tanda lain termasuk gegaran di tangan dan kedutan otot di lidah.
Gejala Atrofi Otot Tunjang Juvana Jenis 3
Atrofi otot tunjang jenis 3 biasanya muncul selepas usia 18 bulan dan sebelum usia 18 tahun. Kanak-kanak yang terjejas boleh duduk, berdiri dan berjalan secara bebas. Walau bagaimanapun, kelemahan otot, terutama pada otot pelvis dan kaki, menyebabkan kiprah berjalan.
Selama beberapa tahun, prestasi menurun: Pada mulanya, mereka yang terjejas sukar melakukan aktiviti sukan atau menaiki tangga, tetapi akhirnya sukar juga membawa beg belanja, misalnya. Setelah bertahun-tahun, atrofi otot tulang belakang jenis 3 menjadikan berlari dan aktiviti lain sukar atau bahkan mustahil, walaupun pada orang tua.
Namun, secara keseluruhan, gejala kurang jelas daripada dua bentuk penyakit lain, jenis 1 dan jenis 2. Bagi kebanyakan mereka yang terkena, kualiti hidup hampir tidak terganggu dalam jangka masa yang panjang.
Gejala atrofi otot tulang belakang dewasa jenis 4
Bentuk pembaziran otot progresif yang sangat jarang berlaku bermula pada masa dewasa, selalunya dari dekad ketiga kehidupan. Otot kaki dan pinggul pada mulanya terjejas. Ketika penyakit ini berkembang, kelemahan otot juga merebak ke bahu dan lengan.
Manifestasi gambaran klinikal serupa dengan jenis SMA remaja 3. Walau bagaimanapun, kelemahan otot progresif bahkan lebih lambat daripada dengan jenis SMA 3.
Apa yang menyebabkan atrofi otot tulang belakang?
Pada atrofi otot tulang belakang, neuron motor kedua di saraf tunjang binasa. Ini adalah sel saraf yang mengawal otot dengan pelengkap mereka. Akibat kerosakan pada neuron motor yang sangat khusus ini, lebih sedikit isyarat elektrik yang sampai ke otot daripada yang berlaku pada orang yang sihat. Sekiranya sel otot digunakan kurang dan oleh itu kurang dirangsang, tubuh akan memecahnya dari masa ke masa.
Kecacatan genetik
Dalam kebanyakan kes, atrofi otot tulang belakang adalah penyakit keturunan (SMA keturunan). Penyebab bentuk SMA proksimal tipikal adalah maklumat yang tidak betul dalam susunan genetik pesakit. Gen SMN1 yang disebut pada kromosom 5 tidak berfungsi.
Gen SMN1 membawa maklumat - iaitu cetak biru - untuk molekul protein penting yang disebut SMN. SMN bermaksud "Survival (of) Motor Neuron". Tanpa molekul protein SMN, neuron motor binasa dari masa ke masa.
Memang benar bahawa ada juga gen SMN2 yang berkaitan di dalam tubuh, yang pada prinsipnya dapat "mengimbangi" maklumat genetik SMN1 yang tidak berfungsi. Tetapi ini biasanya hanya berlaku sedikit. Ini bermaksud bahawa kehilangan fungsi gen SMN1 (jika tidak dirawat) biasanya tidak dapat dikompensasikan sepenuhnya oleh salinan gen SMN2 yang utuh.
Pewarisan autosomal resesif dan autosomal dominan
Maklumat genetik seseorang boleh didapati dalam rangkap dua. Hasilnya, setiap orang mempunyai dua salinan gen SMN1 - satu dari bapa mereka dan satu dari ibu mereka. Atrofi otot tulang belakang proksimal pada masa kanak-kanak biasanya diwarisi sebagai sifat resesif autosomal.
Ini bermaksud bahawa kedua-dua varian gen (alel) dari ibu bapa mesti cacat agar atrofi otot tulang belakang berkembang pada keturunan. Sekiranya berlaku pusaka resesif, ibu bapa tidak terjejas kerana, selain yang tidak berfungsi, mereka juga mempunyai gen SMN1 yang sihat yang mengimbangi kecacatan tersebut.
Di sekitar setiap orang ke-45 adalah pemilik sistem ini untuk SMA. Pasangan di mana kedua-dua pasangan adalah pembawa mempunyai risiko 25% mempunyai anak dengan penyakit ini.
Dalam beberapa kes pada masa remaja, atrofi otot tulang belakang pada masa dewasa khususnya juga mengikuti warisan dominan autosomal. Sekiranya berlaku warisan yang dominan, gen yang rosak sudah menyatakan diri - dan mereka yang terjejas menjadi sakit. Walau bagaimanapun, ini bukan kerosakan genetik yang disebutkan di atas kromosom 5. SMA yang berkaitan dengan 5q ini selalu diwarisi secara resesif autosomal.
Warisan dengan bentuk SMA lain
Atrofi otot tulang belakang yang tidak proksimal juga boleh diwarisi. Bentuk spinobulbar khas (jenis Kennedy) diwariskan secara perlahan melalui kromosom seks, kromosom X (ini mempengaruhi varian gen yang mengandungi cetak biru untuk laman dok untuk hormon seks lelaki). Dalam kes bentuk sporadis, warisan tidak dijamin. Tidak banyak yang diketahui di sini mengapa betul-betul neuron motor kedua binasa.
Pemeriksaan dan diagnosis
Diagnosis atrofi otot tulang belakang biasanya dibuat oleh pakar pediatrik, pakar pediatrik yang pakar dalam penyakit saraf (pakar neuropediatrik) dan pakar penyakit sistem saraf (pakar neurologi). Pelbagai ujian diperlukan untuk penjelasan yang lebih tepat. Dalam kes SMA, ujian genetik dan pemeriksaan saraf dan otot sangat penting.
Koleksi sejarah perubatan (anamnesis)
Dengan setiap penyakit, doktor terlebih dahulu bertanya mengenai gejala yang telah berlaku dan bagaimana ia berkembang sejauh ini. Pada bayi dan balita, ibu bapa melaporkan perubahan dan kelainan dalam tingkah laku anak mereka. Dalam kes penyakit keturunan khususnya, doktor juga memfokuskan pada sejarah perubatan keluarga.
Pemeriksaan fizikal
Pada dasarnya, doktor menentukan kelainan dalam perkembangan motor dengan memeriksa anak secara fizikal. Sebagai contoh, ia menguji sama ada kanak-kanak boleh memegang kepala secara tegak, duduk atau menggerakkan lengan atau kaki secara bebas (bergantung pada usia mereka).
Ujian latihan pelengkap dilakukan pada kanak-kanak dan orang dewasa yang lebih tua dengan disyaki atrofi otot tulang belakang. Doktor memeriksa berapa banyak kekuatan yang dapat dikumpulkan oleh orang yang bersangkutan dan berapa lama dia dapat menahannya. Dia juga menguji ketahanan.
Di samping itu, doktor menguji refleks, yang biasanya lemah atau padam, terutama dalam kes atrofi otot tulang belakang yang jelas. Untuk melakukan ini, dia mengetuk pelbagai tendon dengan tukul, misalnya di tumit atau di bawah lutut, dan memeriksa reaksinya.
Kajian Genetik
Kaedah pengesanan yang paling boleh dipercayai untuk (keturunan) atrofi otot tulang belakang adalah analisis genetik. Doktor mencari bukti gen SMN1 yang diubah (bermutasi) dan jumlah salinan SMN2 yang ada.
Peraturan umum adalah untuk mendiagnosis dan merawat (keturunan) SMA seawal mungkin. Bergantung pada bentuk dan rawatan yang ada, perkembangan motor dapat dipengaruhi secara positif sebelum neuron motorik saraf tunjang mengalami kerosakan yang tidak dapat dipulihkan.
Siasatan lanjut di SMA
Sekiranya SMA disyaki, doktor sering mengukur kelajuan konduksi saraf (elektroneurografi) dan aktiviti otot (elektromiografi). Sekiranya perlu, mereka juga memeriksa otot menggunakan ultrasound (myosonography) atau pencitraan resonans magnetik (MRI).
Di samping itu, doktor mengatur ujian darah. Sekiranya terdapat atrofi otot tulang belakang, parameter tertentu dapat diubah: sebagai contoh, tahap kreatin kinase (CK, enzim otot khas) meningkat.
Rawatan atrofi otot tulang belakang
Rawatan atrofi otot tulang belakang adalah kompleks. Untuk masa yang lama, terapi kausal tidak mungkin dilakukan untuk sebarang bentuk SMA. Walau bagaimanapun, kemajuan dalam penyelidikan perubatan memberi doktor pilihan rawatan baru untuk asasnya membantu mereka yang terkena SMA proksimal (kecacatan gen SMN pada kromosom 5).
Di samping itu, doktor menumpukan perhatian untuk meredakan gejala dan memberikan sokongan terbaik kepada mereka yang terjejas (misalnya fisioterapi, terapi pernafasan, psikoterapi, mungkin pembedahan).
Terapi perubatan
Pendekatan rawatan baru untuk pesakit di mana SMA berdasarkan kecacatan gen SMN yang diketahui campur tangan secara langsung dalam bahan genetik itu sendiri atau dalam pemprosesan hiliran maklumat genetik.
Tujuannya adalah untuk membolehkan tubuh pesakit menghasilkan kuantiti protein SMN yang mencukupi, yang sangat penting untuk neuron motor.
Pilihan rawatan berikut tersedia untuk atrofi otot tulang belakang:
- Modulator penyambungan (Nusinersen, Risdiplam): Ubat-ubatan ini campur tangan secara langsung dalam pemprosesan molekul RNA messenger selanjutnya. Mereka menguatkan proses yang memberikan jumlah protein SMN yang lebih tinggi dari gen SMN2 yang utuh.
- Terapi penggantian gen (Onasemnogene Abeparvovec): Terapi ini campur tangan secara langsung dalam genom manusia. Salinan gen SMN1 yang salah diganti di sel yang terjejas oleh pembinaan gen berfungsi yang dibekalkan secara luaran.
Modulator penyambungan
Sekiranya terdapat kecacatan gen SMN1, tubuh secara alternatif dapat menghasilkan protein SMN dari gen SMN2 yang berkaitan. Gen pengganti SMN2 "melompat", tetapi itu tidak mencukupi. Sebabnya: Protein di SMN2 biasanya terlalu pendek dan dipecah dengan cepat.
Ini disebabkan oleh pemprosesan RNA pemesejan SMN2 yang sesuai (SMN2 mRNA). Ia menghantar maklumat pembinaan dari genom (DNA) ke lokasi pengeluaran protein (ribosom).
Untuk melakukan ini, gen SMN2 dalam genom pertama kali dibaca. RNA utusan SMN2 awal dihasilkan. Antara lain, ia mesti diproses lebih jauh melalui penyambungan yang disebut. Barulah RNA utusan dewasa muncul. Kompleks sel khas, ribosom, kemudian membaca RNA utusan dewasa dan dengan itu menghasilkan protein SMN2. Dan justru inilah yang dipendekkan dan tidak stabil, cepat dibongkar dan tidak dapat mengambil alih fungsi SMN1.
Untuk mengubahnya, bahan aktif Nusinersen dan Risdiplam mempengaruhi pemprosesan selanjutnya RNA utusan awal. Akibatnya, modulator penyambungan yang disebut ini akhirnya meningkatkan jumlah protein SMN yang dapat digunakan - dan dengan itu dapat memastikan bekalan yang mencukupi.
Nusinersen
Ubat Nusinersen adalah apa yang disebut "antisense oligonucleotide" (ASO). Ia diluluskan oleh Agensi Ubat Eropah pada tahun 2017. ASO dihasilkan molekul RNA buatan dan disesuaikan khas. Mereka mengikat secara khusus dan tepat pada RNA utusan SMN2. Ini menghalang mereka daripada diproses secara tidak betul dalam sel manusia.
Khususnya: Nusinersen menghalang maklumat penting (exon 7) daripada tersalah memotong RNA pemesejan SMN2. Keberadaan exon 7 menyebabkan tubuh seterusnya menghasilkan protein SMN yang lebih berfungsi.
Nusinersen diberikan melalui apa yang dikenali sebagai tusukan lumbal. Ini bermaksud bahawa ubat itu disuntik ke saluran tulang belakang dengan jarum suntik. Terapi ini diulang secara berkala selama beberapa bulan. Pada tahun pertama rawatan, mereka yang terkena menerima enam, kemudian tiga dos setahun.
Pesakit biasanya bertolak ansur dengan ubat tersebut. Nusinersen membawa kepada kursus penyakit yang lebih baik. Kajian menunjukkan bahawa mobiliti meningkat pada banyak pesakit: dalam banyak kes, adalah mungkin untuk duduk bebas dan memusingkan badan secara bebas. Kesan sampingan dan komplikasi berdasarkan, antara lain, pada tusukan lumbal (mis. Sakit kepala, jangkitan pada meninges).
Risdiplam
Suruhanjaya Eropah meluluskan Risdiplam sebagai ubat ketiga terhadap SMA yang berkaitan dengan 5q (jenis 1-3 atau satu hingga empat salinan gen SMN2) pada bulan Mac 2021. Risdiplam diambil setiap hari sebagai serbuk larut. Dos yang tepat dikira berdasarkan umur dan berat badan.
Tidak seperti Nusinersen, Risdiplam bukanlah "antisense oligonucleotide", tetapi molekul kecil. Molekul ini mengikat pada RNA messenger untuk protein SMN2 dan menstabilkannya dengan cara ini. Hasilnya, protein SMN yang lebih berfungsi dihasilkan.
Kesan sampingan Risdiplam yang biasa termasuk ketidakselesaan gastrousus, ruam, demam, dan jangkitan saluran kencing.
Terapi penggantian gen
Pendekatan lain untuk rawatan atrofi otot tulang belakang proksimal bergantung pada apa yang dikenali sebagai terapi penggantian gen. Gen SMN1 yang cacat - titik permulaan SMA progresif - "digantikan" oleh salinan gen berfungsi baru.
Bahan aktif Onasemnogene Abeparvovec (AVXS-101), yang berfungsi berdasarkan prinsip ini, mendapat kebenaran pemasaran bersyarat untuk rawatan balita dan kanak-kanak dari Agensi Ubat Eropah (EMA) pada Mei 2020.
Ubat ini boleh digunakan untuk SMA jenis 1 mengikut maklumat EMA. Dalam semua bentuk penyakit SMA yang lain, ciri genetik (jumlah salinan SMN2) memutuskan sama ada terapi penggantian gen adalah pilihan.
Dengan Onasemnogene Abeparvovec, salinan gen SMN1 manusia diperkenalkan ke dalam sel-sel saraf tunjang dan batang otak yang terjejas. Ini dilakukan oleh virus tertentu yang berfungsi sebagai "feri" untuk bahan genetik baru - yang disebut vektor virus yang berkaitan dengan adeno (vektor AAV).
Konstruksi gen vektor diberikan sekali sebagai infusi melalui vena ke dalam aliran darah dan dari sana disebarkan ke seluruh badan. Oleh kerana penghalang darah-otak yang belum berkembang sepenuhnya pada anak-anak kecil, vektor ini juga dapat masuk ke dalam tisu saraf tunjang.
Dengan mengikat vektor ini dengan struktur permukaan khas neuron motor, mereka memilih bahan genetik untuk menghasilkan protein SMN secara bebas.
Rawatan dapat meningkatkan fungsi motor dan membawa kepada kejayaan perkembangan yang berkekalan (mis. Duduk, merangkak dan berjalan tanpa sokongan).Semasa rawatan, nilai hati kadang-kadang dapat meningkat dengan ketara, tetapi jumlah platelet darah dapat menurun. Demam dan muntah juga biasa. Untuk mengurangkan kesan sampingan, pesakit diberi kortikosteroid ("kortison") selama beberapa minggu.
Perkembangan motorik yang sesuai dengan usia umumnya hanya mungkin dilakukan sekiranya terapi gen telah dimulakan secara pramatang. Rawatan berlaku di pusat rawatan neuromuskular khusus.
terapi fizikal
Fisioterapi terus menjadi tonggak penting dalam rawatan SMA. Tidak semua bentuk SMA dapat dirawat dengan pendekatan rawatan baru. Terapi senaman secara berkala dirancang untuk mengekalkan kemampuan fizikal dan melambatkan kerosakan otot.
Fisioterapis secara pasif menggerakkan bahagian badan yang sudah lumpuh. Urutan pergerakan aktif, pada gilirannya, dilatih untuk menyokong pergerakan dan kekuatan otot. Rawatan urut atau panas dan sejuk juga dapat membantu. Ini juga berfungsi untuk berehat dan, dalam keadaan tertentu, memperlambat kemerosotan selanjutnya.
Terapi ucapan
Dalam beberapa kes, SMA mempengaruhi otot bercakap dan menelan. Kemudian latihan terapi pertuturan membantu. Ini mendorong kanak-kanak belajar bercakap. Walaupun pada pesakit tua, ini biasanya dapat memperlambat kemerosotan pertuturan. Ahli terapi pertuturan juga melatih menelan yang betul.
Kedua-dua ahli fisioterapi dan ahli terapi pertuturan menyokong mereka yang terkena terapi pernafasan yang disasarkan.
Rawatan melegakan kesakitan
Terapi kesakitan memainkan peranan penting, terutama pada peringkat penyakit yang lebih maju. Doktor menggunakan ubat penghilang rasa sakit untuk mengurangkan penderitaan mereka yang terjejas.
pembedahan
Oleh kerana atrofi otot tulang belakang boleh menyebabkan kelengkungan tulang belakang yang teruk (scoliosis), doktor kadang-kadang menganggap pembedahan. Dengan berbuat demikian, mereka secara khusus mengeras tulang belakang.
Ini memberi kestabilan batang tubuh (yang tertentu) yang terjejas, yang bukan hanya membolehkan postur yang lebih tegak, tetapi juga melindungi tulang dan sendi. Pembedahan tulang belakang juga dapat membantu mengatasi masalah pernafasan progresif.
Penjagaan psikoterapi
Penyakit neuromuskular seperti atrofi otot tulang belakang mewakili tekanan psikologi yang hebat.Pesakit dan pesakit memproses diagnosis dalam sesi individu dan kumpulan yang dipimpin oleh psikoterapi dan mengembangkan strategi untuk menangani penyakit ini dengan lebih baik.
Kumpulan pertolongan diri dan wakil pesakit juga menawarkan sokongan penting. Mereka memberi maklumat, memberi nasihat dan menyokong mereka yang terkena dampak dan saudara-mara mereka untuk menghadapi cabaran penyakit SMA.
Kemungkinan pemulihan dari atrofi otot tulang belakang
Sekiranya terdapat atrofi otot tulang belakang, prognosis bergantung terutamanya pada bentuk masing-masing. Semakin lama muncul gejala, semakin baik jalannya. Sebagai tambahan, doktor yang lebih awal mendiagnosis atrofi otot tulang belakang, lebih cepat mereka dapat melakukan langkah-langkah rawatan yang sesuai, bahkan sebelum neuron motorik mengalami kerosakan yang tidak dapat dipulihkan.
Pilihan rawatan baru melalui modulator penyambungan dan terapi penggantian gen berpotensi besar dalam rawatan SMA proksimal - terutama ketika memulakan rawatan (sangat) lebih awal. Walau bagaimanapun, data untuk prognosis jangka panjang yang dipercayai masih belum selesai. Hanya kajian lanjutan dan pemerhatian keselamatan ubat-ubatan yang dekat dapat memberikan kepastian lebih lanjut dalam beberapa bulan ke depan. Dengan ubat-ubatan yang lebih baru, kawalan penyakit jangka panjang atau bahkan penawarnya paling tidak dapat dibayangkan.
SMA jenis 1 pada amnya adalah penyakit yang serius. Kanak-kanak yang menghidap SMA jenis 1 mempunyai (tidak dirawat) jangka hayat yang sangat terhad. Kelemahan otot yang cepat meningkat di seluruh badan juga mempengaruhi pernafasan. Akibatnya adalah radang paru-paru akut dan bahkan kegagalan pernafasan. Kanak-kanak yang terjejas mati dalam beberapa tahun pertama kehidupan.
Prognosis sedikit lebih baik untuk SMA jenis 2. Jangka hayat berbeza-beza bergantung pada keparahan penyakit yang tepat: ada yang mati pada masa kanak-kanak, tetapi kebanyakannya mencapai usia dewasa. Cepat atau lambat - jika dikehendaki - pernafasan mesti disokong dalam bentuk yang lebih teruk. Orang yang terjejas tetap bergerak dengan bantuan kerusi roda.
Dengan SMA jenis 3, prognosisnya jauh lebih baik - terutamanya jika gejala pertama muncul lewat. Prestasi secara beransur-ansur merosot selama beberapa tahun. Pada usia tua, kerusi roda atau penjagaan tetap mungkin diperlukan. Jangka hayat hampir tidak dibatasi oleh atrofi otot tulang belakang jenis 3.
Atrofi otot tulang belakang dewasa (jenis 4) lebih perlahan daripada jenis 3. Orang biasanya mempunyai jangka hayat yang normal.
Tags.: nilai makmal ubatan alternatif diet