Phenylketonuria
Marian Grosser belajar perubatan manusia di Munich. Di samping itu, doktor, yang berminat dalam banyak perkara, berani membuat jalan memutar yang menarik: mempelajari falsafah dan sejarah seni, bekerja di radio dan, akhirnya, juga untuk seorang Netdoctor.
Lebih banyak mengenai pakar Semua kandungan diperiksa oleh wartawan perubatan.Phenylketonuria (PKU) adalah penyakit keturunan, metabolisme protein keturunan. Ia menghalang pemecahan fenilalanin asid amino. Ini berkumpul di dalam badan dan mengganggu perkembangan otak anak. Sekiranya tidak dirawat, fenilketonuria menyebabkan kecacatan intelektual yang teruk. Dengan terapi tepat pada masanya, pesakit dapat menjalani kehidupan yang normal. Dapatkan maklumat mengenai phenylketonuria di sini!
Kod ICD untuk penyakit ini: Kod ICD adalah kod yang diiktiraf di peringkat antarabangsa untuk diagnosis perubatan. Mereka boleh didapati, misalnya, dalam surat doktor atau sijil ketidakupayaan untuk bekerja. E70
Phenylketonuria: keterangan
Phenylketonuria (PKU) adalah penyakit metabolik yang diwarisi yang wujud sejak lahir dan mengganggu pemecahan fenilalanin asid amino penting. Asid amino adalah asas asas protein dan oleh itu komponen metabolik penting. Sebilangan dari mereka hanya dapat masuk ke dalam tubuh melalui makanan, organisma tidak dapat menghasilkannya sendiri. Asid amino seperti itu disebut mustahak.
Apa yang berlaku dengan fenilketonuria?
Biasanya, asid amino mengalami keseimbangan antara penyerapan / penumpukan dan pemecahan, sehingga selalu tersedia sebanyak yang diperlukan oleh tubuh. Sekiranya terdapat pelbagai asid amino, kekurangan atau kelebihan boleh menyebabkan bahaya dan menyebabkan pelbagai gejala.
Dalam PKU klasik yang disebut, kesan fenilalanin hidroksilase (PAH) adalah terhad atau bahkan tidak ada sama sekali. Kerana kekurangan PAH, fenilalanin semakin berkumpul di dalam badan. Kepekatan fenilalanin yang terlalu tinggi mengganggu perkembangan otak dan menyebabkan kecacatan intelektual pada pesakit muda pada peringkat awal.
Kerana pemecahan normal fenilalanin tidak mungkin berlaku dengan penyakit ini, produk pemecahan lain, yang disebut fenol keton, terbentuk. Mereka dikeluarkan dalam air kencing dan bertanggungjawab untuk nama penyakit ini.
Fenilketonuria atipikal
Walaupun dengan bentuk fenilketonuria yang tidak biasa, pemecahan fenilalanin terganggu. Namun, penyebabnya bukanlah kecacatan pada PAH. Sebaliknya, fungsi koenzim, tetrahydrobiopterin (BH4), dihalang. Ia secara tidak langsung terlibat dalam pemecahan fenilalanin kerana PAH memerlukan BH4 untuk menukar fenilalanin menjadi tirosin.
Kerana BH4 juga penting untuk penghasilan zat utusan dopamin dan serotonin, fenilketonuria atipikal dengan kekurangan BH4 biasanya lebih rumit daripada bentuk klasik.
Siapa yang mempengaruhi phenylketonuria?
PKU adalah salah satu penyakit metabolik kongenital yang paling biasa. Dianggarkan bahawa sekitar satu daripada 7,000 bayi baru lahir di seluruh dunia akan mengembangkannya, tanpa perbezaan antara gadis dan lelaki. Oleh kerana ia adalah penyakit keturunan, beberapa anggota keluarga sering terkena.
Phenyketonuria: gejala
Pada mulanya, bayi dengan fenilketonuria tidak menunjukkan sebarang gejala penyakit. Masalah pertama tidak berlaku sehingga bulan keempat hingga keenam kehidupan, jika penyakit ini belum dikenali dan dirawat. Yang paling penting, pematangan otak yang terganggu menyebabkan komplikasi besar dari masa ke masa. Gejala PKU yang tidak dirawat termasuk:
- defisit mental yang kuat. Kerosakan otak berlaku melalui akil baligh dan kemudian berhenti. Kanak-kanak yang terjejas biasanya cacat mental.
- Kejang (sawan) (epilepsi). Kerana kerosakan, sel-sel saraf otak sangat sensitif dan berlebihan. Kejang epilepsi yang kerap adalah hasilnya.
- kecacatan motor. Bukan hanya sel-sel otak, tetapi juga otot-otot pesakit yang terlalu teruja. Itulah sebabnya ia sering menjadi tegang (spasticity), yang menyebabkan pelbagai gangguan pergerakan.
- Gangguan tingkah laku. Beberapa kanak-kanak dengan fenilketonuria hiperaktif dan agresif yang luar biasa, dan kemarahan juga sering berlaku.
- kepala kecil (microcephaly). Kerana otak pesakit tidak berkembang dengan baik, pertumbuhan kepala juga tertinggal. Lingkaran kepala yang kecil berbanding dengan rakan sebaya mereka sangat ketara pada kanak-kanak yang lebih tua.
- bau yang ketara. PKU menghasilkan produk pemecahan fenilalanin tertentu yang berbau seperti kotoran tikus. Bahan-bahan ini terutama dikeluarkan di dalam air kencing, tetapi juga sebahagiannya melalui kulit.
- perubahan kulit seperti ekzema
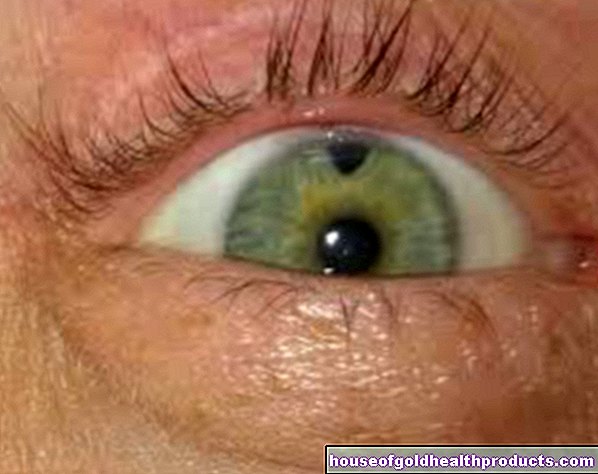
Kerana pengeluaran pigmen melanin juga terganggu dalam fenilketonuria, banyak penderita mempunyai kulit yang sangat ringan, sensitif terhadap sinar matahari dan rambut berambut putih. Mata iris juga berwarna biru muda hingga telus dan membolehkan fundus kemerahan bersinar.
Gejala PKU bervariasi dalam tahap keparahan dari orang ke orang. Sebab utama untuk ini adalah bahawa aktiviti fenilalanin hidroksilase (PAH) dibatasi secara berbeza pada setiap pesakit. Sebilangan masih mempunyai aktiviti sisa tertentu, sehingga kurang fenilalanin terkumpul di dalam organisma. Yang lain sama sekali tidak menunjukkan aktiviti enzim - penyakit ini berkembang lebih pantas dan lebih serius.
Phenylketonuria: penyebab dan faktor risiko
Phenylketonuria adalah penyakit keturunan. Banyak mutasi genetik kini diketahui yang menyebabkan kecacatan PAH. Jenis mutasi menentukan sejauh mana pemecahan fenilalanin dibatasi.
PKU diwarisi secara resesif, yang bermaksud bahawa seseorang boleh menjadi pembawa gen yang diubah tanpa mengembangkan penyakit. Begitu juga, orang yang mempunyai fenilketonuria dapat melahirkan anak yang sihat.
Hanya jika kedua ibu bapa mempunyai mutasi dalam susunan genetik mereka, ada kemungkinan bahawa keturunan mereka akan mengembangkan fenilketonuria. Sekiranya kedua ibu bapa bukan sahaja menjadi pembawa gen, tetapi juga mempunyai PKU sendiri, semua anak bersama-sama juga akan mengembangkannya.
Phenylketonuria: pemeriksaan dan diagnosis
Oleh kerana akibat serius dari fenilketonuria dapat dicegah dengan memulakan rawatan pada waktunya, sangat penting untuk mengetahui penyakit ini seawal mungkin. Di Jerman, anak-anak diperiksa untuk pelbagai penyakit kongenital, termasuk PKU, sebagai bagian dari pemeriksaan umum (pemeriksaan bayi baru lahir) pada hari ketiga setelah kelahiran.
Spektrometri jisim tandem
Banyak gangguan metabolik kongenital kini didiagnosis dengan bantuan spektrometri jisim tandem yang disebut. Ia membolehkan darah bayi yang baru lahir diperiksa dengan cepat dan mudah. Selain fenilketonuria, doktor dapat mengesan lebih daripada 20 penyakit lain dalam beberapa minit.
Ujian Guthrie
Ujian Guthrie, dinamai penemuinya, juga memungkinkan untuk mendiagnosis PKU. Untuk melakukan ini, sejumlah kecil darah diambil dari tumit anak dan disapu pada sehelai kertas turas. Di makmal anda kemudian dapat menentukan sama ada kepekatan fenilalanin meningkat.
Ujian Guthrie diperkenalkan pada tahun 1960-an dan telah lama menjadi kaedah standard untuk mendiagnosis fenilketonuria. Walau bagaimanapun, ia mempunyai kelemahan berbanding dengan spektrometri jisim tandem. Ujian Guthrie hanya memberikan hasil setelah lima hari, yang juga terdedah kepada kesalahan. Sebagai contoh, faktor seperti diet anak atau kemungkinan terapi antibiotik memalsukan hasilnya. Di beberapa negara ujian Guthrie masih digunakan, di Jerman biasanya tidak lagi digunakan.
Ujian lain
Sekiranya pemeriksaan bayi baru lahir menunjukkan kecurigaan fenilketonuria, pemeriksaan selanjutnya akan disahkan. Ini juga dapat digunakan untuk menentukan kepekatan fenilalanin yang tepat dalam darah.
Akhirnya, adalah penting untuk membezakan sama ada PKU khas (klasik) atau atipikal. Ujian khas juga disediakan untuk ini, seperti ujian tekanan tetrahidrobiopterin. Perbezaannya penting kerana fenilketonuria atipikal diperlakukan secara berbeza daripada bentuk klasik.
Pemeriksaan cecair amniotik
Phenylketonuria sudah boleh didiagnosis semasa kehamilan (diagnosis pranatal). Untuk melakukan ini, sejumlah kecil cecair amniotik diambil dari kantung ketuban ibu dan sel-sel anak yang belum lahir yang terkandung di dalamnya diperiksa. Sebarang kecacatan genetik yang menyebabkan PKU dapat dikenal pasti dengan cara ini.
Dengan pemeriksaan bayi yang baru lahir, phenylketonuria biasanya dijumpai cukup awal untuk dirawat. Oleh kerana ujian cecair amniotik selalu dikaitkan dengan risiko tertentu, penggunaannya untuk mendiagnosis PKU biasanya tidak berguna.
Phenylketonuria: Rawatan
Hanya ada satu cara untuk mengatasi kelebihan fenilalanin di PKU: Anak-anak yang terjejas mesti mengikuti diet khas untuk mengambil makanan fenilalanin sesedikit mungkin dengan makanan mereka. Mereka juga memerlukan makanan tambahan untuk menggantikan zat yang akan terbentuk dari fenilalanin pada anak yang sihat.
Terapi mesti dimulakan sebelum gejala pertama gangguan perkembangan muncul, iaitu dalam dua bulan pertama kehidupan. Kerosakan otak yang telah berlaku tidak dapat dibalikkan.
Terapi pemakanan dengan diet PKU
Semua protein semula jadi terdiri daripada kira-kira lima peratus fenilalanin asid amino. Sebilangan besar makanan mengandungi lebih banyak daripada yang diperlukan oleh badan. Ini tidak menjadi masalah bagi orang yang sihat kerana dia memecah dan mengeluarkan jumlah fenilalanin yang berlebihan.
Pesakit dengan fenilketonuria, sebaliknya, harus melakukan tanpa protein semula jadi. Produk khas yang dikeluarkan secara industri menggantikan komponen makanan yang hilang. Di satu pihak, seseorang ingin mencegah kelebihan fenilalanin, di sisi lain, pesakit harus dibekalkan dengan bahan-bahan yang mungkin kekurangannya, seperti tirosin, yang terbentuk dari fenilalanin.
Walau bagaimanapun, tujuan diet PKU bukanlah untuk menghentikan penyerapan fenilalanin sepenuhnya. Kerana organisma memerlukan sejumlah asid amino untuk proses metabolik yang penting. Mencapai tumpuan yang betul menimbulkan cabaran besar bagi doktor dan pesakit dan memerlukan banyak disiplin.
Oleh itu, diet mesti dipatuhi seumur hidup, tetapi sangat ketat sehingga usia enam tahun. Kerana otak berkembang sangat kuat hingga usia ini dan oleh itu sangat mudah mengalami kerosakan. Pada masa dewasa, kepekatan fenilalanin yang tinggi tidak menyebabkan kerosakan otak seperti yang berlaku pada kanak-kanak. Walau bagaimanapun, mereka boleh menjadi pencetus kepada aduan neurologi lain seperti kepekatan yang lemah atau reaksi yang perlahan.
Rawatan diet untuk fenilketonuria harus bermula di pusat penyakit metabolik pakar. Kerana tidak mungkin di setiap klinik untuk memberi petunjuk kepada orang tua mengenai diet. Ini paling baik dilakukan dengan bantuan kaunseling diet, yang menunjukkan cara menjalani diet dan memantau tahap fenilalanin darah secara berkala.
Rawatan fenilketonuria atipikal
Dalam bentuk PKU yang tidak biasa, koenzim BH4 yang hilang dan bahan utusan tertentu seperti dopamin dan serotonin diganti secara buatan. Dalam beberapa kes, pesakit juga mesti menjalani diet rendah fenilalanin.
Phenylketonuria Semasa Kehamilan
Sekiranya anda mengandung dan mengalami fenilketonuria sendiri, anda harus mempertimbangkan perkara berikut:
- Ikuti diet anda dengan ketat!
- Pastikan nilai darah fenilalanin anda diperiksa secara berkala. Kerana kepekatan fenilalanin pada anak yang belum lahir kira-kira dua kali lebih tinggi daripada wanita hamil. Selain otak, mereka juga boleh merosakkan jantung dan mata anak yang belum lahir. Kecacatan pada kerangka anak juga boleh timbul dari tahap fenilalanin yang tinggi semasa kehamilan.
- Untuk mengelakkan kerosakan otak pada anak pada awal kehamilan, wanita dengan fenilketonuria harus merancang kehamilan dengan berhati-hati dan mengambil langkah berjaga-jaga sejak awal.
- Pada wanita hamil dengan fenilketonuria, diet yang tidak dipatuhi dengan ketat boleh menyebabkan keguguran (pengguguran spontan).
- Walau apa pun, wanita hamil dengan fenilketonuria harus mendapatkan nasihat dan bimbingan daripada doktor.
Phenylketonuria: kursus penyakit dan prognosis
Sekiranya fenilketonuria didiagnosis lebih awal, jika mungkin pada bayi baru lahir, dan jika diet PKU khas diikuti, prognosis biasanya baik. Anak-anak berkembang secara rohani dan mempunyai jangka hayat rata-rata.
Namun, jika tidak dirawat, kerosakan otak menyebabkan gangguan perkembangan mental yang teruk yang tidak dapat diperbaiki kemudian. Mereka yang terjejas mempunyai jangka hayat yang normal, tetapi hasil kecerdasan mereka hampir selalu jauh di bawah norma.
Bentuk atipikal fenilketonuria yang jarang berlaku, di mana terdapat kekurangan BH4, adalah kes khas. Varian fenilketonuria ini boleh menyebabkan kerosakan neurologi progresif dengan kekejangan teruk walaupun diet.
Tags.: rawatan rumah pertolongan cemas remaja